Las enfermedades de depósito lisosomal (EDL) conforman un grupo de enfermedades altamente diverso. El tratamiento inicial es general y de soporte. Como son enfermedades multisistémicas, un enfoque multidisciplinario es esencial; es posible que muchos especialistas distintos participen en el cuidado de los pacientes. Se recomienda la participación temprana de los centros especializados; los cuidados deben estar coordinados por el centro. En el caso de muchos pacientes, no hay un tratamiento específico (p. ej., enfermedad de Gaucher neonatal tipo 2) y es posible que la mejor opción sea un tratamiento paliativo.
La terapia de reposición enzimática (TRE) se ha establecido para varias de las EDL.[3]Mehta A, Winchester B, eds. Lysosomal storage disorders: a practical guide. 2nd ed. Oxford: Wiley-Blackwell; 2022. En Europa, la TRE puede administrarse en el propio domicilio del paciente para los trastornos de Gaucher, Fabry, Pompe, mucopolisacaridosis (MPS) I y MPS II.
Fisioterapeutas, ergoterapeutas, especialistas en enfermería clínica, psicólogos, docentes y trabajadores sociales; todos ellos pueden formar parte del extenso equipo multidisciplinario.
Enfermedades de Gaucher
Enfermedad de Gaucher tipo 1
El retraso en el diagnóstico sigue siendo motivo de preocupación y es necesario aumentar la concienciación de los médicos generales, pediatras y hematólogos.[98]Kishnani PS, Al-Hertani W, Balwani M, et al. Screening, patient identification, evaluation, and treatment in patients with Gaucher disease: results from a Delphi consensus. Mol Genet Metab. 2022 Feb;135(2):154-62.
https://www.sciencedirect.com/science/article/pii/S109671922101194X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/34972655?tool=bestpractice.com
Las personas levemente afectadas, por ejemplo, con hemoglobina normal, plaquetas >100 x 10^9/L, agrandamiento mínimo del hígado y del bazo y sin lesiones óseas en la resonancia magnética (IRM), requieren observación pero pueden no necesitar un tratamiento específico. El tratamiento de pacientes asintomáticos no está justificado.[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58.
http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com
Debe evitarse la esplenectomía; mejora los hemogramas pero predispone a enfermedades óseas más graves a largo plazo y a hipertensión pulmonar.[100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86.
https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x
http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com
Puede producirse enfermedad esquelética que derive en dolor óseo o fracturas; la cirugía ortopédica puede ser necesaria para tratar las articulaciones afectadas.
En caso de haber una mayor incidencia de neoplasias malignas hematológicas en adultos, se les debe monitorizar para detectar: en particular, mieloma, pero también otras neoplasias como el linfoma no Hodgkin.[100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86.
https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x
http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com
La enfermedad de Parkinson y la cirrosis hepática son otras complicaciones a largo plazo que requieren monitorización y tratamiento.
Deben buscarse y tratarse las anomalías endocrinas y metabólicas coexistentes, como la resistencia a la insulina, la deficiencia de la hormona del crecimiento y la insuficiencia de vitamina D.[101]Kałużna M, Trzeciak I, Ziemnicka K, et al. Endocrine and metabolic disorders in patients with Gaucher disease type 1: a review. Orphanet J Rare Dis. 2019 Dec 2;14(1):275.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-019-1211-5
http://www.ncbi.nlm.nih.gov/pubmed/31791361?tool=bestpractice.com
Está disponible la TRE con imiglucerasa, velaglucerasa alfa y taliglucerasa alfa.[102]Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010 Jun 10;115(23):4651-6.
http://www.bloodjournal.org/content/115/23/4651.long
http://www.ncbi.nlm.nih.gov/pubmed/20299511?tool=bestpractice.com
[103]Morris JL. Velaglucerase alfa for the management of type 1 Gaucher disease. Clin Ther. 2012 Feb;34(2):259-71.
http://www.ncbi.nlm.nih.gov/pubmed/22264444?tool=bestpractice.com
[104]Ben Turkia H, Gonzalez DE, Barton NW, et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am J Hematol. 2013 Mar;88(3):179-84.
http://www.ncbi.nlm.nih.gov/pubmed/23400823?tool=bestpractice.com
[105]Hughes DA, Gonzalez DE, Lukina EA, et al. Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: long-term data from phase III clinical trials. Am J Hematol. 2015 Jul;90(7):584-91.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4654249
http://www.ncbi.nlm.nih.gov/pubmed/25801797?tool=bestpractice.com
La presencia de TRE debe considerarse en todos los niños sintomáticos con la enfermedad de Gaucher, y en adultos con reducciones significativas en los hemogramas (p. ej., nivel de hemoglobina <100 g/L [10 g/dL], plaquetas <100 x 10⁹/L), agrandamiento significativo de los órganos (p. ej., tamaño del bazo >10 veces el normal), la presencia de enfermedad esquelética demostrada en la IRM y/o cualquier otro daño orgánico (p. ej., evidencia de daño pulmonar).[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58.
http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com
[100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86.
https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x
http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com
Se ha demostrado el beneficio de la TRE en la mejora de las alteraciones hematológicas, del dolor óseo y en la reducción del tamaño del hígado y del bazo. La densidad ósea, la función pulmonar y la calidad de vida también mejoran.[106]Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency - macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991 May 23;324(21):1464-70.
http://www.ncbi.nlm.nih.gov/pubmed/2023606?tool=bestpractice.com
[107]Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type I Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002 Aug 1;113(2):112-9.
http://www.ncbi.nlm.nih.gov/pubmed/12133749?tool=bestpractice.com
[108]Gabrowski GA, Kolodny EH, Weinreb NJ, et al. Gaucher disease: phenotypic and genetic variation. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The metabolic and molecular basis of inherited disease. 9th ed. New York, NY: McGraw-Hill; 2006.
Se ha demostrado que la terapia de reducción de sustrato (TRS) mejora la anemia, la trombocitopenia, el agrandamiento de hígado/bazo y la osteoporosis.[109]Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015 Feb 17;313(7):695-706.
http://jama.jamanetwork.com/article.aspx?articleid=2110969
http://www.ncbi.nlm.nih.gov/pubmed/25688781?tool=bestpractice.com
La TRS (por ejemplo, miglustat, eliglustat) es un tratamiento por vía oral que se emplea en pacientes con intolerancia a la TRE o con acceso venoso difícil.[109]Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015 Feb 17;313(7):695-706.
http://jama.jamanetwork.com/article.aspx?articleid=2110969
http://www.ncbi.nlm.nih.gov/pubmed/25688781?tool=bestpractice.com
[110]Cox TM, Aerts JM, Andria G, et al. The role of the iminosugar N-butyldeoxynorjirimycin (miglustat) in the management of type 1 (non-neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis. 2003;26(6):513-26.
http://www.ncbi.nlm.nih.gov/pubmed/14605497?tool=bestpractice.com
[111]Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000 Apr 29;355(9214):1481-5.
http://www.ncbi.nlm.nih.gov/pubmed/10801168?tool=bestpractice.com
[112]Pastores GM, Giraldo P, Cherin P, et al. Goal-oriented therapy with miglustat in Gaucher disease. Curr Med Res Opin. 2009 Jan;25(1):23-37.
http://www.ncbi.nlm.nih.gov/pubmed/19210136?tool=bestpractice.com
[113]Poole RM. Eliglustat: first global approval. Drugs. 2014 Oct;74(15):1829-36.
http://www.ncbi.nlm.nih.gov/pubmed/25239269?tool=bestpractice.com
Se han reconocido los objetivos de la terapia para la enfermedad de Gaucher tipo 1 y los tratamientos disponibles actualmente permiten lograr la mayoría de ellos.[114]Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: an expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis. 2018 Feb;68:203-8.
https://www.sciencedirect.com/science/article/pii/S1079979616301917?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/28274788?tool=bestpractice.com
Se han realizado trasplantes de células madre, pero la TRE es más segura.[115]Peters C, Steward CG, National Marrow Donor Program, et al. Hematopoietic cell transplantation for inherited metabolic disorders: an overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003 Feb;31(4):229-39.
http://www.ncbi.nlm.nih.gov/pubmed/12621457?tool=bestpractice.com
Enfermedad de Gaucher tipo 2
La enfermedad de Gaucher tipo 2 neuronopática aguda solo debe tratarse mediante cuidados de soporte, ya que la muerte prematura es frecuente.
Las complicaciones de la enfermedad son convulsiones, retraso en el desarrollo neurológico y trastornos del movimiento ocular y deben considerarse parte del manejo de la enfermedad.
La enfermedad de Gaucher tipo 2 es prácticamente intratable.
La terapia de reposición enzimática no es eficaz.
Enfermedad de Gaucher tipo 3
Los trastornos del movimiento ocular, el retraso en el desarrollo neurológico y la enfermedad esquelética son complicaciones de esta enfermedad.
Normalmente afecta a niños y adultos jóvenes; los aspectos viscerales y esqueléticos de la enfermedad responden bien a la TRE, pero las manifestaciones neurológicas no mejoran, puesto que la TRE no puede atravesar la barrera hematoencefálica.[116]Vellodi A, Tylki-Szymanska A, Davies EH, et al. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis. 2009 Oct;32(5):660-4.
http://www.ncbi.nlm.nih.gov/pubmed/19655269?tool=bestpractice.com
Enfermedad de Fabry
El manejo de esta enfermedad multisistémica incluye un gran número de aspectos de soporte generales.
El alivio del dolor con gabapentina o pregabalina está indicado para el dolor neuropático. El uso de carbamazepina también está muy extendido.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
[117]Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018 Jul;124(3):189-203.
http://www.ncbi.nlm.nih.gov/pubmed/30017653?tool=bestpractice.com
Los antiinflamatorios no esteroideos se deben utilizar con moderación, ya que con frecuencia estos pacientes presentan nefropatía.
Las lesiones cutáneas pueden requerir cirugía estética o terapia con láser.[Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) fosa lumbar, (B) genitales, (C) ombligo, (D) zona lumbar, (E) dedos de los piesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].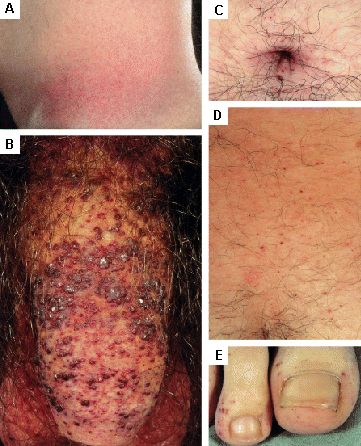 [Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].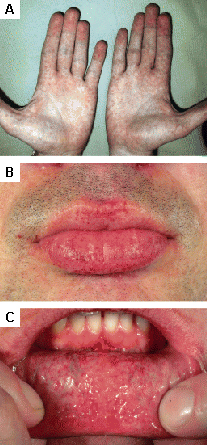
La presión arterial y la proteinuria se deben monitorizar periódicamente, con una intervención temprana mediante inhibidores de la enzima convertidora de la angiotensina (IECA) o antagonistas de los receptores de la angiotensina II.[118]Jain G, Warnock DG. Blood pressure, proteinuria and nephropathy in Fabry disease. Nephron Clin Pract. 2011;118(1):43-8.
http://www.ncbi.nlm.nih.gov/pubmed/21071972?tool=bestpractice.com
La revisión cardíaca es esencial; el reconocimiento temprano de la arritmia es importante. Deben considerarse todas las posibilidades quirúrgicas, incluida la colocación de marcapasos, la resección del tabique, el reemplazo de válvulas e incluso el trasplante cardíaco.[119]Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage diseases. Heart. 2007 Apr;93(4):528-35.
http://www.ncbi.nlm.nih.gov/pubmed/17401074?tool=bestpractice.com
Los accidentes cerebrovasculares y los ataques isquémicos transitorios requieren cuidadosas medidas de prevención primaria y secundaria.
La evaluación gastrointestinal, la terapia sintomática y endoscopia pueden ser necesarias para excluir enfermedades relacionadas.[120]Hoffmann B, Schwarz M, Mehta A, et al. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007 Dec;5(12):1447-53.
http://www.ncbi.nlm.nih.gov/pubmed/17919989?tool=bestpractice.com
La depresión es frecuente en la enfermedad de Fabry; cuando sea posible, debe observarse a la familia en conjunto. Es posible que sea necesario ofrecer asesoramiento.
La sordera es altamente probable.[121]Germain DP, Avan P, Chassaing A, et al. Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty-two hemizygous male patients. BMC Med Genet. 2002 Oct 11;3:10.
https://bmcmedgenet.biomedcentral.com/articles/10.1186/1471-2350-3-10
http://www.ncbi.nlm.nih.gov/pubmed/12377100?tool=bestpractice.com
Tratamiento específico con terapia de reposición enzimática (TRE) o terapia con chaperonas
Las directrices para el diagnóstico y el tratamiento están disponibles y se actualizan periódicamente.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
[117]Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018 Jul;124(3):189-203.
http://www.ncbi.nlm.nih.gov/pubmed/30017653?tool=bestpractice.com
[122]El Dib R, Gomaa H, Carvalho RP, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2016 Jul 25;(7):CD006663.
http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006663.pub4/full
http://www.ncbi.nlm.nih.gov/pubmed/27454104?tool=bestpractice.com
[123]Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019 Aug;96(2):107-17.
https://onlinelibrary.wiley.com/doi/10.1111/cge.13546
http://www.ncbi.nlm.nih.gov/pubmed/30941742?tool=bestpractice.com
[124]Germain DP, Altarescu G, Barriales-Villa R, et al. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol Genet Metab. 2022 Sep-Oct;137(1-2):49-61.
https://www.sciencedirect.com/science/article/pii/S1096719222003705?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35926321?tool=bestpractice.com
Generalmente se reconoce que la TRE puede ralentizar el avance del daño orgánico en los riñones y el corazón, pero es posible que dichos órganos no recuperen su función normal. Los hombres deben tratarse tan pronto como se les diagnostique; las mujeres deben tratarse si presentan síntomas de afectación de órganos importantes.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
El inicio de la TRE debe considerarse en pacientes femeninas asintomáticas con evidencia de laboratorio, histológica o de imagen de afectación renal, cardíaca o del sistema nervioso central.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
La agalsidasa alfa y la agalsidasa beta han demostrado su eficacia y seguridad en ensayos aleatorizados controlados.[125]Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N Engl J Med. 2001 Jul 5;345(1):9-16.
http://www.nejm.org/doi/full/10.1056/NEJM200107053450102#t=article
http://www.ncbi.nlm.nih.gov/pubmed/11439963?tool=bestpractice.com
[126]Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001 Jun 6;285(21):2743-9.
http://jama.jamanetwork.com/article.aspx?articleid=193887
http://www.ncbi.nlm.nih.gov/pubmed/11386930?tool=bestpractice.com
Los ensayos y los datos de los registros han demostrado la reducción del riesgo de accidente cerebrovascular, la mejora de los resultados cardíacos y renales, la mejora del dolor y la calidad de vida, y la mejora de la sintomatología gastrointestinal.[127]Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008 Feb;94(2):153-8.
http://www.ncbi.nlm.nih.gov/pubmed/17483124?tool=bestpractice.com
[128]Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007 Jan 16;146(2):77-86.
http://www.ncbi.nlm.nih.gov/pubmed/17179052?tool=bestpractice.com
[129]Beck M, Ricci R, Widmer U, et al. Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest. 2004 Dec;34(12):838-44.
http://www.ncbi.nlm.nih.gov/pubmed/15606727?tool=bestpractice.com
[130]Mehta A, Beck M, Elliott P, et al; Fabry Outcome Survey investigators. Enzyme replacement therapy with agalsidase alfa in patients with Fabry's disease: an analysis of registry data. Lancet. 2009 Dec 12;374(9706):1986-96.
http://www.ncbi.nlm.nih.gov/pubmed/19959221?tool=bestpractice.com
[131]Germain DP, Arad M, Burlina A, et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease - a systematic literature review by a European panel of experts. Mol Genet Metab. 2019 Mar;126(3):224-35.
https://www.sciencedirect.com/science/article/pii/S1096719218301926
http://www.ncbi.nlm.nih.gov/pubmed/30413388?tool=bestpractice.com
[132]Sheng S, Wu L, Nalleballe K, et al. Fabry's disease and stroke: effectiveness of enzyme replacement therapy (ERT) in stroke prevention, a review with meta-analysis. J Clin Neurosci. 2019 Jul;65:83-6.
http://www.ncbi.nlm.nih.gov/pubmed/30955952?tool=bestpractice.com
[133]Spada M, Baron R, Elliott PM, et al. The effect of enzyme replacement therapy on clinical outcomes in paediatric patients with Fabry disease - a systematic literature review by a European panel of experts. Mol Genet Metab. 2019 Mar;126(3):212-23.
https://www.sciencedirect.com/science/article/pii/S1096719218301860
http://www.ncbi.nlm.nih.gov/pubmed/29785937?tool=bestpractice.com
Estos fármacos son seguros para el uso en niños y presentan el mismo beneficio en hombres y mujeres. La TRE provoca la producción de anticuerpos en hombres, pero se desconoce el impacto de esto en la eficacia del tratamiento. Las mujeres no desarrollan anticuerpos, quizás porque son heterocigóticas y presentan enzima circulante. Tanto la agalsidasa alfa como la agalsidasa beta son eficaces para prevenir las complicaciones renales y cardiovasculares, en comparación con la ausencia de tratamiento. La agalsidasa beta se asocia a un menor riesgo de complicaciones cerebrovasculares, en comparación con la agalsidasa beta o la ausencia de tratamiento.[134]El Dib R, Gomaa H, Ortiz A, et al. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS One. 2017 Mar 15;12(3):e0173358.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0173358
http://www.ncbi.nlm.nih.gov/pubmed/28296917?tool=bestpractice.com
La TRE es eficaz para reducir las puntuaciones de dolor y las concentraciones de globotriaosilceramida en plasma, riñón y corazón.[122]El Dib R, Gomaa H, Carvalho RP, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2016 Jul 25;(7):CD006663.
http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006663.pub4/full
http://www.ncbi.nlm.nih.gov/pubmed/27454104?tool=bestpractice.com
La pegunigalsidasa alfa es otra opción para tratar la enfermedad de Fabry en adultos y tiene la ventaja de una vida media plasmática más larga.[135]National Institute for Health and Care Excellence. Pegunigalsidase alfa for treating Fabry disease. Oct 2023 [internet publication].
https://www.nice.org.uk/guidance/ta915
La terapia con chaperonas consiste en pequeñas moléculas que potencian la actividad de la enzima deficiente en pacientes con actividad enzimática residual.[136]Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471-86.
http://www.ncbi.nlm.nih.gov/pubmed/25587658?tool=bestpractice.com
Estos compuestos ayudan al tráfico intracelular de la enzima para mejorar el suministro al lisosoma desde el retículo endoplasmático. Puede funcionar en combinación con la terapia de reposición enzimática, puesto que no se ve afectada por anticuerpos.
Migalastat es una terapia con una chaperona oral que aumenta la actividad de la enzima endógena alfa-galactosidasa A en pacientes con una mutación susceptible. Los ensayos han demostrado la seguridad y utilidad de esta terapia en pacientes con mutaciones susceptibles, y el tratamiento está ahora autorizado y disponible en Estados Unidos, Europa, Canadá, Japón y muchos otros países.[137]Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016 Aug 11;375(6):545-55.
http://www.nejm.org/doi/full/10.1056/NEJMoa1510198
http://www.ncbi.nlm.nih.gov/pubmed/27509102?tool=bestpractice.com
[138]National Institute for Health and Care Excellence. Migalastat for treating Fabry disease. Feb 2017 [internet publication].
https://www.nice.org.uk/guidance/hst4
[139]Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017 Apr;54(4):288-96. [Erratum in: J Med Genet. 2018.]
https://jmg.bmj.com/content/54/4/288.long
http://www.ncbi.nlm.nih.gov/pubmed/27834756?tool=bestpractice.com
[140]Schiffmann R, Bichet DG, Jovanovic A, et al. Migalastat improves diarrhea in patients with Fabry disease: clinical-biomarker correlations from the phase 3 FACETS trial. Orphanet J Rare Dis. 2018 Apr 27;13(1):68.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0813-7
http://www.ncbi.nlm.nih.gov/pubmed/29703262?tool=bestpractice.com
Los médicos deben confirmar que la mutación del paciente es susceptible. El fabricante recomienda que se evite si la tasa de filtración glomerular estimada (TFGe) es inferior a 30 mL/minuto/1.73 m². El migalastat es adecuado tanto para los pacientes nativos como para los que cambian de ERT.[141]Bichet DG, Hopkin RJ, Aguiar P, et al. Consensus recommendations for the treatment and management of patients with Fabry disease on migalastat: a modified Delphi study. Front Med (Lausanne). 2023 Sep 1:10:1220637.
https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1220637/full
http://www.ncbi.nlm.nih.gov/pubmed/37727761?tool=bestpractice.com
Enfermedades de mucopolisacaridosis (MPS)
Manejo general y de soporte
Puesto que la MPS es una enfermedad multisistémica, es necesario un enfoque multidisciplinario. Es preciso obtener pronto asesoramiento de un especialista.
Los primeros problemas pueden surgir con enfermedad cardíaca y respiratoria; en todo momento debe prestarse especial atención a las vías respiratorias.[142]Shinhar SY, Zablocki RN, Madgy DN. Airway management in mucopolysaccharide disorders. Arch Otolaryngol. 2004 Feb;130(2):233-7.
http://www.ncbi.nlm.nih.gov/pubmed/14967758?tool=bestpractice.com
Las complicaciones de oído, nariz y garganta incluyen incluyen frecuentes infecciones de oído.[143]Simmons MA, Bruce IA, Penney S, et al. Otorhinological manifestations of mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2005 May;69(5):589-95.
http://www.ncbi.nlm.nih.gov/pubmed/15850680?tool=bestpractice.com
Las complicaciones musculoesqueléticas son frecuentes, así que es esencial contar con el asesoramiento de otros especialistas, como cirujanos ortopédicos y neurocirujanos.[144]van der Linden MH, Kruyt MC, Sakkers RJ, et al. Orthopaedic management of Hurler's disease after hematopoietic stem cell transplantation: a systematic review. J Inherit Metab Dis. 2011 Jun;34(3):657-69.
http://link.springer.com/article/10.1007/s10545-011-9304-x/fulltext.html
http://www.ncbi.nlm.nih.gov/pubmed/21416194?tool=bestpractice.com
[145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24.
http://www.sciencedirect.com/science/article/pii/S1096719212002740
http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com
Una complicación potencialmente mortal, sobre todo durante la anestesia, es la compresión de la médula espinal debido a estenosis en la región craneocervical.[145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24.
http://www.sciencedirect.com/science/article/pii/S1096719212002740
http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com
El síndrome del túnel carpiano es frecuente, al igual que las deformidades de la columna.[146]Pastores GM, Meere PA. Musculoskeletal complications associated with lysosomal storage disorders: Gaucher disease and Hurler-Scheie syndrome (mucopolysaccharidosis type I). Curr Opin Rheumatol. 2005 Jan;17(1):70-8.
http://www.ncbi.nlm.nih.gov/pubmed/15604908?tool=bestpractice.com
Se debe realizar una evaluación cardíaca previa a la cirugía en el caso de todos los pacientes, dado el número de alteraciones valvulares que se dan en este grupo de trastornos.
Tratamiento específico
Se debe considerar el trasplante de células madre para aquellos pacientes afectados de gravedad; su valor está demostrado, por ejemplo, en MPS I y MPS VI graves.[147]Boelens JJ. Trends in hematopoietic stem cell transplantation for inborn errors of metabolism. J Inherit Metab Dis. 2006 Apr-Jun;29(2-3):413-20.
http://www.ncbi.nlm.nih.gov/pubmed/16763911?tool=bestpractice.com
La ERT es de beneficio establecido en las MPS I, II, IVA (síndrome de Morquio A), VI y tipo VII (síndrome de Sly).[148]Hendriksz CJ, Burton B, Fleming TR, et al.; STRIVE Investigators. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014 Nov;37(6):979-90.
http://link.springer.com/article/10.1007/s10545-014-9715-6/fulltext.html
http://www.ncbi.nlm.nih.gov/pubmed/24810369?tool=bestpractice.com
[149]Brunelli MJ, Atallah ÁN, da Silva EM. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis type VI. Cochrane Database Syst Rev. 2021 Sep 17;(9):CD009806.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009806.pub3/full
http://www.ncbi.nlm.nih.gov/pubmed/34533215?tool=bestpractice.com
[150]Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2019 Jun 18;6(6):CD009354.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009354.pub5/full
http://www.ncbi.nlm.nih.gov/pubmed/31211405?tool=bestpractice.com
[151]Wikman-Jorgensen PE, López Amorós A, Peris García J, et al. Enzyme replacement therapy for the treatment of Hunter disease: a systematic review with narrative synthesis and meta-analysis. Mol Genet Metab. 2020 Sep - Oct;131(1-2):206-10.
http://www.ncbi.nlm.nih.gov/pubmed/32773276?tool=bestpractice.com
[152]Gomes DF, Gallo LG, Leite BF, et al. Clinical effectiveness of enzyme replacement therapy with galsulfase in mucopolysaccharidosis type VI treatment: systematic review. J Inherit Metab Dis. 2019 Jan;42(1):66-76.
http://www.ncbi.nlm.nih.gov/pubmed/30740728?tool=bestpractice.com
Hay varias enzimas aprobadas para estas indicaciones. En el Reino Unido, el National Institute for Health and Care Excellence recomienda la elosulfasa alfa como opción para el tratamiento de la MPS IVA en personas de todas las edades, y solo está disponible bajo un acuerdo comercial.[153]National Institute for Health and Care Excellence. Elosulfase alfa for treating mucopolysaccharidosis type 4A: highly specialised technologies guidance. Apr 2022 [internet publication].
https://www.nice.org.uk/guidance/hst19
Se están realizando ensayos de fase 3 con vestronidasa alfa.[154]NHS; National Institute for Health Research (NIHR). NIHR Innovation Observatory evidence briefing. Vestronidase alfa (UX-003) for mucopolysaccharidosis type VII (MPS 7; Sly syndrome) NIHRIO (HSRIC) ID: 11463. Apr 2017 [internet publication].
http://www.io.nihr.ac.uk/wp-content/uploads/migrated_new/11463-Vestronidase-alfa-UX-003.pdf
[155]ClinicalTrials.gov. A study of UX003 recombinant human beta-glucuronidase (rhGUS) enzyme replacement therapy in subjects with mucopolysaccharidosis type 7, Sly syndrome (MPS 7). Jul 2020 [internet publication].
https://clinicaltrials.gov/ct2/show/NCT02432144
[156]Akyol MU, Alden TD, Amartino H, et al. Recommendations for the management of MPS VI: systematic evidence- and consensus-based guidance. Orphanet J Rare Dis. 2019 May 29;14(1):118.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-019-1080-y
http://www.ncbi.nlm.nih.gov/pubmed/31142378?tool=bestpractice.com
Enfermedad de Pompe
Los cuidados generales y de soporte de neonatos son multidisciplinarios y engloban a neurólogos, anestesistas y cardiólogos. La terapia de reposición enzimática (TRE) para niños y adultos está autorizada.[157]Stevens D, Milani-Nejad S, Mozaffar T. Pompe disease: a clinical, diagnostic, and therapeutic overview. Curr Treat Options Neurol. 2022 Nov;24(11):573-88.
https://link.springer.com/article/10.1007/s11940-022-00736-1
http://www.ncbi.nlm.nih.gov/pubmed/36969713?tool=bestpractice.com
Las revisiones sistemáticas no han identificado ningún ensayo que compare la eficacia y seguridad de la terapia de reposición enzimática para la enfermedad de Pompe de inicio infantil con otra intervención, ninguna intervención o placebo.[158]Chen M, Zhang L, Quan S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst Rev. 2017 Nov 20;(11):CD011539.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD011539.pub2/full
http://www.ncbi.nlm.nih.gov/pubmed/29155436?tool=bestpractice.com
[159]Joanne M, Skye N, Tracy M. The effectiveness of enzyme replacement therapy for juvenile-onset Pompe disease: a systematic review. J Inherit Metab Dis. 2019 Jan;42(1):57-65.
https://onlinelibrary.wiley.com/doi/10.1002/jimd.12027
http://www.ncbi.nlm.nih.gov/pubmed/30740732?tool=bestpractice.com
Un ensayo de 18 participantes comparó dos dosis de alglucosidasa alfa. El ensayo proporcionó evidencia de baja calidad de que el tratamiento con alglucosidasa alfa a largo plazo prolongó notablemente la supervivencia, así como la supervivencia sin ventilación, y mejoró la cardiomiopatía (evidencia de baja calidad); pero la función cardíaca, el desarrollo motor y la proporción de niños sin ventilación invasiva fueron similares para ambas dosis durante una duración media del tratamiento de 2.3 años. No se observaron diferencias significativas entre los grupos de dosis.[160]Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009 Sep;66(3):329-35.
https://www.nature.com/articles/pr2009210
http://www.ncbi.nlm.nih.gov/pubmed/19542901?tool=bestpractice.com
Se necesitan ensayos aleatorizados con potencia adecuada para determinar el régimen de dosis óptimo.
La TRE para la enfermedad de Pompe de inicio tardío se asocia con una mejoría significativa de la distancia recorrida, pero no de la fuerza muscular ni de la capacidad vital forzada.[161]Sarah B, Giovanna B, Emanuela K, et al. Clinical efficacy of the enzyme replacement therapy in patients with late-onset Pompe disease: a systematic review and a meta-analysis. J Neurol. 2021 Apr 13 [Epub ahead of print].
https://link.springer.com/article/10.1007%2Fs00415-021-10526-5
http://www.ncbi.nlm.nih.gov/pubmed/33851281?tool=bestpractice.com
Se necesitan más ensayos prospectivos. La avalglucosidasa es una opción para la enfermedad de Pompe de inicio tardío en niños de 1 año o más.[162]Kishnani PS, Diaz-Manera J, Toscano A, et al. Efficacy and safety of avalglucosidase alfa in patients with late-onset Pompe disease after 97 weeks: a phase 3 randomized clinical trial. JAMA Neurol. 2023 Jun 1;80(6):558-67.
https://jamanetwork.com/journals/jamaneurology/fullarticle/2802973#google_vignette
http://www.ncbi.nlm.nih.gov/pubmed/37036722?tool=bestpractice.com
[163]Dalmia S, Sharma R, Ramaswami U, et al. Enzyme replacement therapy for late-onset Pompe disease. Cochrane Database Syst Rev. 2023 Dec 12;12(12):CD012993.
http://www.ncbi.nlm.nih.gov/pubmed/38084761?tool=bestpractice.com
[164]National Institute for Health and Care Excellence. Cipaglucosidase alfa with miglustat for treating late-onset Pompe disease. Aug 2023 [internet publication].
https://www.nice.org.uk/guidance/ta912
La cipaglucosidasa alfa es otra opción que está aprobada para el tratamiento de la enfermedad de Pompe de inicio tardío en adultos que no están mejorando su TRE actual. La cipaglucosidasa alfa solo está aprobada para el uso en combinación con miglustat (un estabilizador enzimático).[165]Blair HA. Cipaglucosidase alfa: first approval. Drugs. 2023 Jun;83(8):739-45.
https://link.springer.com/article/10.1007/s40265-023-01886-5
http://www.ncbi.nlm.nih.gov/pubmed/37184753?tool=bestpractice.com
Las guías de práctica clínica de consenso europeas recomiendan un período inicial de 2 años de TRE para los adultos sintomáticos con la enfermedad de Pompe. La TRE puede continuar durante el embarazo y la lactancia. La función muscular y respiratoria debe evaluarse durante el tratamiento.[166]van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017 Jun;24(6):768-e31.
http://www.ncbi.nlm.nih.gov/pubmed/28477382?tool=bestpractice.com
enfermedad de Tay-Sachs
Los cuidados paliativos solo se requieren en la forma infantil de la enfermedad.
Las formas de inicio juvenil y las formas crónicas o de inicio en la edad adulta requieren cuidados de soporte, evaluación de necesidades de educación especiales y evaluación neurológica. La demencia y la ataxia son complicaciones a largo plazo que se deben considerar en el manejo.
La terapia de reposición enzimática (TRE) no está disponible para la enfermedad de Tay-Sachs.
Enfermedades de Niemann-Pick
Solo se requieren cuidados paliativos en la forma infantil grave de la enfermedad de Niemann-Pick tipo A. Los cuidados de soporte están indicados para las formas menos graves.
La enfermedad de Niemann-Pick tipo B normalmente se presenta en adultos con enfermedad pulmonar y/o hepatoesplenomegalia. Esta afección es prácticamente intratable. Las terapias de soporte deben estar guiadas por evaluaciones realizadas por un neumólogo, un gastroenterólogo y un hematólogo.[167]Geberhiwot T, Wasserstein M, Wanninayake S, et al. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann-Pick disease types A, B and A/B). Orphanet J Rare Dis. 2023 Apr 17;18(1):85.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-023-02686-6
http://www.ncbi.nlm.nih.gov/pubmed/37069638?tool=bestpractice.com
La enfermedad de Niemann-Pick tipo C es extremadamente variable, pero la evaluación neurológica es generalmente importante. La terapia de reducción de sustrato (TRS) está disponible para la enfermedad de Niemann-Pick tipo C.[54]Patterson MC, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017 Dec;7(6):499-511.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5800709
http://www.ncbi.nlm.nih.gov/pubmed/29431164?tool=bestpractice.com
Se ha demostrado que la SRT con miglustat estabiliza los síntomas neurodegenerativos, incluida la disfagia.[168]Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J Rare Dis. 2018 Aug 15;13(1):140.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0844-0
http://www.ncbi.nlm.nih.gov/pubmed/30111334?tool=bestpractice.com
La evaluación neurológica antes de la TRS es importante.