Abordaje
Las enfermedades de depósito lisosomal (EDL) son un grupo de enfermedades clínicamente diversas, con un vasto diagnóstico diferencial. Las distintas EDL se confunden con facilidad; la derivación a un especialista en una etapa temprana es importante.
En la actualidad existen tratamientos disponibles para muchas EDL y, por lo tanto, hay un énfasis cada vez mayor en la necesidad de un diagnóstico y una intervención tempranos, antes de que se produzcan daños orgánicos importantes.
El retraso del diagnóstico es un problema importante en el caso de todas las EDL. El diagnóstico de una EDL se debe considerar en casos con características clínicas relevantes indicativas de una EDL.
Las EDL son trastornos hereditarios y, por lo tanto, se deben tener en cuenta los antecedentes familiares. Se debe solicitar la colaboración de un genetista clínico si se va a realizar un cribado familiar.
Anamnesis y exploración física
Los diagnósticos clínico y de laboratorio deben integrarse y se debe considerar la posibilidad de una enfermedad de depósito lisosomal (EDL) en aquellos casos que presenten características clínicas relevantes que indiquen una EDL.
Enfermedad de Gaucher
La enfermedad de Gaucher tipo 1 generalmente se presenta en adultos con trombocitopenia y/o esplenomegalia.[30] La fatiga y la hepatomegalia son síntomas frecuentes.[30] Se produce con mayor frecuencia en judíos asquenazíes.[10][11]
La enfermedad de Gaucher (tipo 2 grave, neuronopática aguda) se presenta en neonatos con retraso en el desarrollo, dificultades en la alimentación, hepatoesplenomegalia, piel anormal, convulsiones y enfermedad del sistema nervioso central manifiesta; generalmente, estos pacientes mueren durante los primeros meses de vida.[30]
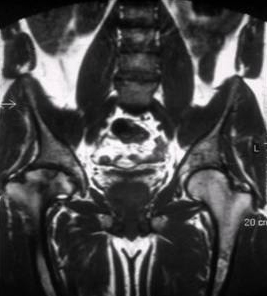
El tipo 3 (neuronopatía crónica) se presenta sobre todo en edades comprendidas entre 10 y 20 años con trastorno del movimiento ocular, hepatoesplenomegalia y dolor óseo. También pueden producirse crisis convulsivas, cataratas, valvulopatía cardíaca, contracturas articulares y depresión.[41][42][Figure caption and citation for the preceding image starts]: Resonancia magnética del esqueleto en la enfermedad de Gaucher tipo 1 que muestra una deposición generalizada de sustrato en el esqueleto con necrosis asociada e infarto óseo. Se ha producido necrosis en la cabeza del fémur (flecha)De la colección personal del Profesor Atul B. Mehta [Citation ends].
Puede producirse parkinsonismo asociado con la enfermedad de Gaucher en la edad adulta.[30]
Puede haber antecedentes inespecíficos de infección recurrente de las vías respiratorias.
Trastornos del movimiento ocular, como la parálisis vertical de la mirada, son frecuentes en la enfermedad infantil.[43]
Enfermedad de Fabry
El diagnóstico de la enfermedad de Fabry a menudo se omite en la infancia.[38]
Las características frecuentes incluyen ardor doloroso en las extremidades, fiebre, dolor abdominal y diarrea.[16][17][44] Puede presentarse en la edad adulta con accidente isquémico transitorio o accidente cerebrovascular, agrandamiento del corazón (cardiomiopatía hipertrófica) e insuficiencia renal crónica.[31]
Pueden producirse opacidad corneal, cataratas, hipertensión, hipotiroidismo, valvulopatía cardíaca, deterioro auditivo/sordera repentina y depresión.[12]
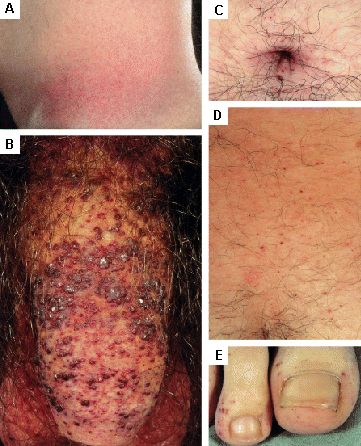
También se observan lesiones cutáneas.[31] Algunos ejemplos son el angioqueratoma y la telangiectasia.[Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) fosa lumbar, (B) genitales, (C) ombligo, (D) zona lumbar, (E) dedos de los piesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].
 [Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].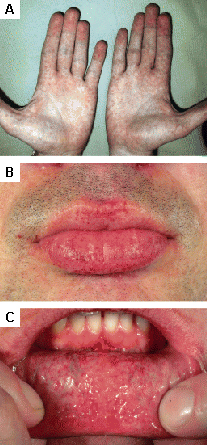
Los síntomas orales incluyen agenesia dental, dientes supernumerarios e hiposalivación.[45]
Con frecuencia (>75% de los casos), las mujeres heterocigóticas son sintomáticas.[36][37][38][39][40]
Mucopolisacaridosis (formas graves de MPS I, II y otras)
Generalmente, la MPS se presenta en el periodo neonatal o en la lactancia/primera infancia con rasgos faciales anormales, circunferencia de la cabeza grande, hidrocefalia, agrandamiento de la lengua, hepatoesplenomegalia, disostosis y malformación espinal (giba incluida), opacidad corneal, retraso en el desarrollo neurológico, deformidad de las articulaciones y retraso en el desarrollo.[5][46]
También pueden producirse alteraciones de las válvulas cardíacas, disnea y dificultades en la deglución.[47] El trastorno cognitivo es frecuente.
Los niños con MPS III pueden presentar síntomas de trastorno del espectro autista, incluyendo retraso en el lenguaje y deterioro de la comunicación social. Los síntomas suelen aparecer a una edad más avanzada que el trastorno del espectro autista idiopático, tras un periodo de desarrollo normal.[48]
Las MPS también pueden presentarse como hidropesía fetal no inmune recurrente.[1]
Puede haber antecedentes inespecíficos de infección recurrente de las vías respiratorias.[49]
Las variantes atenuadas aparecen más tarde (generalmente entre los 5 y los 15 años de edad), pero pueden presentarse en pacientes >30 años con deformidad de las articulaciones, síndrome del túnel carpiano, hepatoesplenomegalia, neuropatía de compresión, cardiopatía y enfermedad pulmonar, y deterioro auditivo/sordera repentina.
Enfermedad de Pompe
También conocida como enfermedad del almacenamiento del glucógeno de tipo II, la forma clásica infantil se presenta en las primeras semanas o meses de vida en forma de dificultades de alimentación, retraso en el desarrollo, hipotonía y agrandamiento del corazón con ECG anormal y cambios ecocardiográficos de la cardiomiopatía hipertrófica.[35][50][51]
Pueden producirse agrandamiento de la lengua, hepatomegalia y deterioro auditivo.[45]
La muerte suele producirse en el primer año de vida sin una terapia específica.
Una característica típica de la enfermedad de Pompe es la fatigabilidad excesiva en todas las edades (p. ej., dificultades en la alimentación durante la lactancia, mal rendimiento deportivo en la infancia, dificultades respiratorias, caídas o dificultad para subir escaleras en la edad adulta).
Puede haber antecedentes inespecíficos de infección recurrente de las vías respiratorias.[52]
Las variantes atenuadas de inicio tardío se presentan entre los 10 y los 20 años (pueden ser tan tardías como >30 años) con síntomas de disfunción del músculo esquelético: dificultad para subir escaleras, levantarse de una posición sentada o acostada, dificultad respiratoria, fatiga, contractura articular y depresión.[53]
enfermedad de Niemann-Pick
La presentación dependerá del tipo: hepatoesplenomegalia en los tipos A, B y C; retraso en el desarrollo neurológico en el tipo A; y demencia progresiva, ataxia o alteración de la marcha, o problemas auditivos en el tipo C.[54][55]
Los síntomas psiquiátricos preceden a los síntomas físicos en el 75% de los pacientes con el tipo C. Las alteraciones cognitivas, de memoria e instrumentales son los síntomas psiquiátricos más comunes, seguidos de la psicosis, la alteración del comportamiento y los trastornos del estado de ánimo.[55]
Los trastornos del movimiento ocular, como la parálisis vertical de la mirada, son frecuentes en el tipo C.[54]
El tipo A se produce con mayor frecuencia en judíos asquenazíes.
enfermedad de Tay-Sachs
En la forma infantil, se presenta con hiperacusia y una mancha macular de color "rojo cereza".[56]
La forma juvenil se presenta con atrofia óptica (retinitis pigmentaria), demencia progresiva y ataxia o alteración de la marcha, retraso en el desarrollo, contractura articular y depresión.
La psicosis, la ataxia, la distonía y la cataplexia en adultos jóvenes pueden ser características de neurodegeneración.[57][58]
Los trastornos del movimiento ocular, como la parálisis vertical de la mirada, son frecuentes.
Se produce con mayor frecuencia en judíos asquenazíes.
El diagnóstico de confirmación se obtiene mediante el análisis de la actividad de la enzima específica o la solicitud de la secuencia genética y estudios familiares. Generalmente lo adecuado es que los generalistas consideren el diagnóstico y lo busquen; no obstante, la derivación temprana a un especialista es recomendable para definir el diagnóstico específico o excluir las EDL en caso de duda.
Análisis enzimático
Es la investigación clave para la mayoría de las enfermedades de depósito lisosomal (EDL). Es posible que sea adecuado solicitar el análisis de una sola enzima (p.ej., glucocerebrosidasa si se sospecha la enfermedad de Gaucher) o de un grupo de enzimas (p. ej., solicitar un análisis enzimático leucocitario como método de cribado de enfermedades de mucopolisacaridosis [MPS]). En las EDL ligadas al cromosoma X (p. ej., enfermedad de Fabry, MPS II), los valores de las mujeres heterocigóticas pueden ser ambiguos, ya que aproximadamente la mitad de las células serán normales.
También es importante solicitar la determinación de la actividad enzimática plasmática además de la actividad leucocitaria, puesto que algunas mutaciones se asocian con una alteración de la exportación enzimática que causa una actividad plasmática inferior con una actividad leucocitaria normal.
Se deben utilizar laboratorios acreditados y especializados, ya que las pruebas son complejas en el aspecto técnico y constantemente se producen ajustes (p. ej., para mejorar el diagnóstico de la enfermedad de Pompe).[59] En la actualidad, hay disponibles directrices para el diagnóstico de laboratorio de la MPS VI.[60] Estas directrices recomiendan precaución en el uso del análisis de glicosaminoglicano (GAG) en orina como único método para confirmar el diagnóstico de la MPS VI y reconocen el análisis de la actividad enzimática como un componente crítico del diagnóstico.
Niveles de sustrato
El aumento de los niveles del sustrato adecuado es detectable en presencia de una deficiencia enzimática. Por lo tanto, los niveles de GAG en orina aumentan en las enfermedades de mucopolisacaridosis (MPS) y los niveles de oligosacáridos aumentan en las gangliosidosis GM1 y GM2.[61] En la enfermedad de Fabry, aumentan los niveles de globotriaosilceramida (Gb3) en orina. Los niveles de tetrasacáridos de glucosa en orina (Glc4) y en plasma (Hex4) están aumentados en los pacientes con la enfermedad de Pompe.[62] Los niveles plasmáticos de glucosilceramida aumentan en la enfermedad de Gaucher.
análisis de ADN
Los estudios genéticos pueden confirmar el diagnóstico de la mayoría de enfermedades de depósito lisosomal (EDL).
En la enfermedad de Gaucher, se observan 6 mutaciones comunes en judíos asquenazíes y un número reducido de otras mutaciones recurrentes de modo que existen pruebas diagnósticas comercializadas para la detección de las mutaciones frecuentes mediante reacción en cadena de la polimerasa (PCR).[63][64] A menudo solo se detecta un alelo mutante; por lo tanto, es preciso considerar las manifestaciones clínicas para diferenciar a los portadores de los enfermos.
En la enfermedad de Fabry, la mayoría de los hombres afectados por la forma clásica presentan mutaciones "particulares" (mutaciones raras que generalmente se encuentran solo en poblaciones pequeñas) que con frecuencia son completas (relacionadas con la enzima ausente).[31]
En la enfermedad de Pompe, las enfermedades de mucopolisacaridosis (MPS) y casi todas las demás EDL, se notifica un gran número de mutaciones para cada enfermedad, pero existen mutaciones recurrentes que normalmente son más comunes en unas comunidades que en otras.
El resultado del análisis de ADN se debe considerar a la luz de la información y los datos clínicos detallados sobre la actividad enzimática.
Algunas mutaciones son polimorfismos y es posible que no estén relacionados con la enfermedad. Muchas pruebas de la PCR solo detectan mutaciones comunes; las mutaciones "particulares" solo se detectan con un análisis secuencial en laboratorios de investigación.
La cromatografía de líquidos desnaturalizante de alta presión es un método para el cribado rápido de mutaciones de base única.
La detección de portadores en trastornos ligados al cromosoma X solo puede lograrse de manera fiable mediante el análisis de ADN.
No todas las mutaciones se han analizado y algunos trastornos solo se diagnostican en laboratorios especializados; es esencial consultar a los centros especializados en cuanto al diseño y la implementación de la estrategia de diagnóstico para estas enfermedades raras.
Hemograma completo
La anemia puede ser multifactorial debido a enfermedad crónica, insuficiencia renal, dificultades en la alimentación o reemplazo de la médula ósea.
Generalmente, la leucopenia se debe a la esplenomegalia. Se pueden observar inclusiones anormales en los leucocitos; por ejemplo, linfocitos ácido periódico de Schiff (PAS) positivos en la enfermedad de Pompe; aspecto anormal de los leucocitos en la sangre/médula ósea en la enfermedad de Tay-Sachs.[65]
La trombocitopenia generalmente se debe a la esplenomegalia, aunque puede darse una alteración de la función plaquetaria, por ejemplo, en el síndrome de Hermansky-Pudlak.[66]
ECG y ecocardiograma
Ambos son esenciales para la evaluación cardíaca en la enfermedad de Pompe infantil, la enfermedad de Fabry en la edad adulta y muchas enfermedades de mucopolisacaridosis (MPS) neonatales.[67][68] En estos pacientes, los hallazgos indicarán agrandamiento de las cavidades cardíacas, válvulas anormales y defectos funcionales.
Pruebas funcionales respiratorias
Son críticas para evaluar la gravedad de la enfermedad de Niemann-Pick tipos A y B. En estos pacientes se detectará una función pulmonar anormal con una transferencia de gases deficiente.[69]
Examen oftálmico
En la enfermedad de Tay-Sachs se observan una mancha macular de "color cereza" y atrofia óptica o retinitis pigmentaria.[56] La opacidad corneal se observa en las enfermedades de mucopolisacaridosis (MPS), la enfermedad de Fabry y algunos tipos de la enfermedad de Gaucher.[43] En la enfermedad de Fabry se observa la catarata característica con vasos retinianos tortuosos.[56] La cataratas también se observan en la enfermedad de Gaucher, las enfermedades de mucopolisacaridosis (MPS) y otras EDL.
Biopsia del tejido afectado
Se debe considerar solo después de haber realizado otras pruebas menos invasivas. La biopsia de tejido de un órgano agrandado y/o con la función alterada (p. ej., hígado, riñón, corazón) puede realizarse para demostrar el aumento de los niveles de sustrato, que a menudo se asocia con congestión celular.
La biopsia de médula ósea a menudo se realiza en la investigación de la hepatoesplenomegalia y puede revelar células de depósito características, como macrófagos cargados de sustrato en la enfermedad de Gaucher.[70] La microscopía electrónica del tejido afectado puede mostrar cambios característicos de daño lisosomal. La biopsia muscular es una herramienta de utilidad diagnóstica en muchas enfermedades de depósito lisosomal (EDL) (p. ej., enfermedad de Pompe de inicio tardío; no obstante, el aspecto puede ser normal); a menudo se prefiere la biopsia de piel, puesto que es relativamente no invasiva y el cultivo de fibroblastos de la piel es de utilidad en el análisis enzimático.[71][72][73] Es el especialista quien debe decidir sobre el tejido adecuado para la biopsia.[Figure caption and citation for the preceding image starts]: Aspirado de médula ósea que muestra una célula de Gaucher típica. Se trata de un macrófago que ha ingerido material celular; el sustrato sin degradar (glucosilceramida) se acumula en los lisosomasDe la colección personal del Profesor Atul B. Mehta [Citation ends].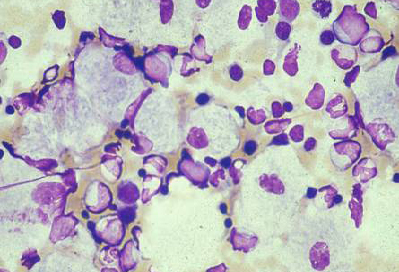 [Figure caption and citation for the preceding image starts]: Imagen de microscopio electrónico de una biopsia de células epiteliales pulmonares en la enfermedad de Fabry que muestra los depósitos de sustrato característicos en los lisosomas, que forman "cuerpos de cebra": (A) ampliación x8000, (B) ampliación x62,500Kelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. Agosto 2000; 55 (8): 720-1; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Imagen de microscopio electrónico de una biopsia de células epiteliales pulmonares en la enfermedad de Fabry que muestra los depósitos de sustrato característicos en los lisosomas, que forman "cuerpos de cebra": (A) ampliación x8000, (B) ampliación x62,500Kelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. Agosto 2000; 55 (8): 720-1; usado con autorización [Citation ends].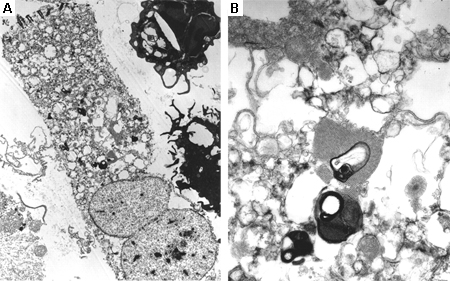
Estudios por imágenes
Las enfermedades de depósito lisosomal son una serie de enfermedades multisistémicas diversas, por lo que son muchas las formas de estudio por imágenes que son importantes y relevantes. Algunos ejemplos son la tomografía computarizada (TC) y la imagen por resonancia magnética (IRM) para medir el volumen de los órganos y evaluar el esqueleto en la enfermedad de Gaucher; el ecocardiograma, la ecografía o la IRM para evaluar el corazón, los riñones y el cerebro en la enfermedad de Fabry; y la TC y las radiografías para evaluar la hidrocefalia comunicante, la displasia atlantoaxoidea y las anomalías espinales/vertebrales en las enfermedades de mucopolisacaridosis (MPS).[42][74][75][76][77][Figure caption and citation for the preceding image starts]: Resonancia magnética del esqueleto en la enfermedad de Gaucher tipo 1 que muestra una deposición generalizada de sustrato en el esqueleto con necrosis asociada e infarto óseo. Se ha producido necrosis en la cabeza del fémur (flecha)De la colección personal del Profesor Atul B. Mehta [Citation ends].
Importancia de los resultados negativos de las pruebas
La derivación temprana a un especialista es necesaria. El médico debe colaborar estrechamente con el laboratorio y garantizar que las muestras llegan rápidamente a este último para evitar su deterioro y la posibilidad de una interpretación falsa negativa. Algunas pruebas están en proceso de mejora (p. ej., para la enfermedad de Pompe); algunas pruebas de ADN/enzimáticas solo están disponibles mediante la derivación a un especialista en laboratorios internacionales de investigación.[78] La deficiencia de una proteína activadora de esfingolípidos o saposina puede producir un fenotipo clínico semejante a una enfermedad de depósito lisosomal (EDL) y puede ser la base de la enfermedad de Gaucher y la leucodistrofia metacromática.[1]
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad