Anamnesis y examen
Principales factores de diagnóstico
común
presencia de factores de riesgo
Los factores de riesgo clave incluyen la etnia asquenazí para muchas enfermedades de depósito lisosomal (EDL) y el sexo masculino para algunas EDL (mucopolisacaridosis II, enfermedad de Fabry). Algunas se presentan en la infancia y otras en la edad adulta. La enfermedad de Gaucher tipo 1 (inicio en la edad adulta), la enfermedad de Niemann-Pick tipo A y la enfermedad de Tay-Sachs son más comunes entre los judíos asquenazíes.
Antecedentes familiares
Estas enfermedades son trastornos hereditarios y se deben evaluar tras la consulta adecuada a un genetista clínico. En ocasiones, los pacientes no presentan antecedentes familiares porque las familias son pequeñas (se trata de trastornos principalmente autosómicos recesivos y los portadores suelen ser asintomáticos) o porque se trata de una nueva mutación. Los antecedentes familiares en el pedigrí ampliado son frecuentes en las enfermedades de depósito lisosomal ligadas al cromosoma X (Fabry, mucopolisacaridosis [MPS] II y Danon).
inicio en la infancia (enfermedades de mucopolisacaridosis [MPS], Pompe, Gaucher, Fabry, Niemann-Pick tipo A)
Edad <1 año: las formas graves de las enfermedades de mucopolisacaridosis (MPS), la enfermedad de Pompe clásica y las formas neuronopáticas de la enfermedad de Gaucher se presentan en la infancia temprana.[30][35][46][50][79] El inicio a una edad temprana es un fuerte indicador de una evolución grave a largo plazo y a menudo se asocia con una actividad enzimática residual baja.
Edad de 1 a 10 años: la mayoría de las MPS (incluidas las variantes atenuadas) y la enfermedad de Niemann-Pick tipo A se presentan en este rango de edad.[80] La mayor parte de los hombres con la enfermedad de Fabry clásica presenta síntomas, aunque la edad media de diagnóstico en hombres es superior.[38]
inicio en la adolescencia (Fabry, Pompe, Gaucher tipos 1, 3, mucopolisacaridosis, Niemann-Pick tipos B, C)
Edad de 10 a 20 años: la mayoría de los casos de la enfermedad de Gaucher tipo 3, la mayoría de los casos, aunque no todos, de la forma más grave de la enfermedad de Gaucher tipo 1, la mayoría de los casos de la enfermedad de Fabry y la mayoría de los casos de la enfermedad de Pompe, incluidos los que se inician en la edad adulta, son sintomáticos, aunque no todas se diagnostican en este rango de edades. Los tipos B y C de la enfermedad de Niemann-Pick son variables; los fenotipos graves se presentan antes y los fenotipos leves aparecen de forma más tardía. Las formas atenuadas de las mucopolisacaridosis también son altamente variables.
inicio en la edad adulta (enfermedades de Fabry, Gaucher tipo 1, Pompe)
Edad de 20 a 50 años: edad típica de inicio para la mayoría de los pacientes con enfermedad de Gaucher tipo 1 y edad a la que los pacientes con la enfermedad de Fabry clásica desarrollan problemas clínicos.
Edad >50 años: es poco probable que las enfermedades por depósito lisosomal se presenten en edades avanzadas; las excepciones son las formas más leves de la enfermedad de Gaucher tipo 1, la enfermedad de Fabry atípica y algunos pacientes con la enfermedad de Pompe más leve.
hepatomegalia y/o esplenomegalia
La hepatomegalia se observa en la enfermedad de Gaucher tipos 2 y 3, las enfermedades de mucopolisacaridosis (MPS), la enfermedad de Pompe y la enfermedad de Niemann-Pick.[5][30][46][49][54]
La esplenomegalia se observa en la enfermedad de Gaucher tipos 2 y 3, las enfermedades de mucopolisacaridosis (MPS) y la enfermedad de Niemann-Pick.[5][30][46][49][54]
La hepatoesplenomegalia es frecuente en las enfermedades de mucopolisacaridosis (MPS), la enfermedad de Gaucher y la enfermedad de Niemann-Pick tipos A, B y C.[5][30][46][49][54]
hiperacusia
Característica de la enfermedad de Tay-Sachs.[81]
antecedentes de insuficiencia renal
Se observan en la enfermedad de Fabry en la edad adulta.[31]
erupción cutánea/lesiones cutáneas
Se encuentra en la enfermedad de Fabry; otras enfermedades de depósito lisosomal.[31][Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) fosa lumbar, (B) genitales, (C) ombligo, (D) zona lumbar, (E) dedos de los piesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].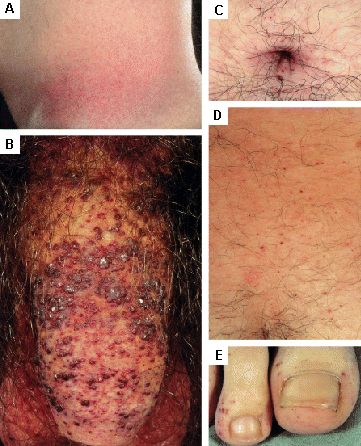 [Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].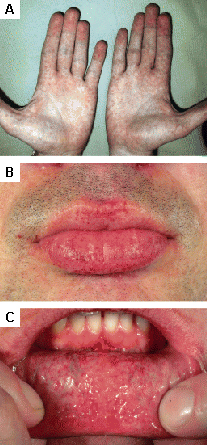
circunferencia de la cabeza grande
mancha macular de color "rojo cereza" en la oftalmoscopia
La coroides vista a través de la fóvea aparece como un área circular roja rodeada de retina de color blanco grisáceo. Es un hallazgo clásico en la enfermedad de Tay-Sachs infantil.[56]
atrofia óptica o retinitis pigmentaria en la oftalmoscopia
En la forma juvenil de la enfermedad de Tay-Sachs se observan atrofia óptica o retinitis pigmentaria.
opacidad corneal en la oftalmoscopia
Otros factores de diagnóstico
común
retraso en el desarrollo neurológico
deterioro auditivo/sordera repentina
Se observa en la enfermedad de Fabry y las enfermedades de mucopolisacaridosis (MPS).[49]
catarata en la oftalmoscopia
En la enfermedad de Fabry se observa la catarata característica con vasos retinianos tortuosos. Las cataratas también se observan en la enfermedad de Gaucher, los trastornos de mucopolisacaridosis y otras enfermedades de depósito lisosomal.[56]
trastorno del movimiento ocular
demencia progresiva y ataxia o alteración de la marcha
Se observa en varias enfermedades de depósito lisosomal juveniles y de inicio en la edad adulta, a menudo acompañadas de ataxia o trastornos de la marcha (p. ej., enfermedad de Tay-Sachs juvenil y crónica, Niemann-Pick tipo C).[54]
retraso en el desarrollo
contractura articular
depresión
Muy frecuente en adultos con la enfermedad de Fabry.[12] También se observa en las enfermedades de Gaucher, Pompe y Tay-Sachs.
alteraciones óseas, incluido giba de la columna
Signo temprano frecuente en las mucopolisacaridosis.[49]
hidrocefalia
Signo temprano frecuente en las mucopolisacaridosis.[49]
antecedentes de infecciones recurrente de las vías respiratorias
psicosis
trastornos del movimiento
infrecuente
accidente cerebrovascular/ataque isquémico transitorio precoz
Observado en la enfermedad de Fabry (frecuentemente circulación posterior).[31]
cardiomegalia
Factores de riesgo
Fuerte
sexo masculino (mucopolisacaridosis [MPS] II, enfermedad de Fabry)
La MPS II es un rasgo ligado al cromosoma X; las manifestaciones en mujeres son extremadamente infrecuentes.
La enfermedad de Fabry también está ligada al cromosoma X, aunque las mujeres heterocigóticas (>75%) normalmente presentan síntomas, si bien son menos graves y más variables en su expresión y aparecen a una edad más avanzada.[36][37][38][39][40]
etnia asquenazí
La tasa aproximada de portadores entre judíos asquenazíes es 1 de cada 15 para la enfermedad de Gaucher, 1 de cada 30 para la enfermedad de Tay-Sachs y entre 1 de cada 80 y 1 de cada 100 para la enfermedad de Niemann-Pick tipo A.[11]
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad