Etiología
Las enfermedades de depósito lisosomal (EDL) son trastornos de un solo gen, generalmente causados por defectos hereditarios en la síntesis (defecto raro en la activación, dirección o tráfico intracelular) de las enzimas lisosómicas.[2]
Estas enzimas son cruciales para la hidrólisis ácida de macromoléculas extracelulares (p. ej., los glicoesfingolípidos derivados de la ruptura de las membranas de las células sanguíneas, los gangliósidos derivados de las neuronas y los glucosaminoglicanos derivados del tejido conectivo y la matriz extracelular). Una cantidad sustancial de sustrato también deriva del recambio intracelular de macromoléculas.
La mayoría de las EDL son autosómicas recesivas; no obstante, 3 de las enfermedades (enfermedad de Fabry, mucopolisacaridosis [MPS] II y enfermedad de Danon) están ligadas al sexo. Existe una heterogeneidad fenotípica considerable entre todas las EDL y las correlaciones genotipo-fenotipo generalmente son escasas. Sin embargo, en general, cuanto menor es la actividad enzimática residual, mayor es la acumulación de sustrato, menor la edad de inicio y más grave la enfermedad.[1][21]
Las deleciones genéticas, las mutaciones sin sentido y las mutaciones que afectan gravemente la estructura terciaria de la enzima generalmente son incompatibles con la vida (p. ej., como se observa en la enfermedad de Gaucher tipo 2) o están relacionadas con manifestaciones graves. Las mutaciones asociadas con la actividad enzimática residual tienen un alto nivel de heterogeneidad fenotípica e incluso hermanos con la misma mutación pueden presentar manifestaciones clínicas muy diferentes.
La afectación neurológica en la enfermedad de Gaucher no se observa en pacientes que tienen al menos un alelo con la mutación N370S (frecuente en los judíos asquenazíes), ya que es suficiente para producir una actividad enzimática que "protege" el sistema nervioso central (SNC).[22][23]
Esfingolipidosis
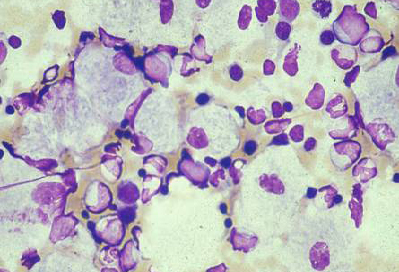
Enfermedad de Gaucher (deficiencia de beta-glucocerebrosidasa) tipo 1 (forma más frecuente), tipos 2 y 3 (con afectación del sistema nervioso central [SNC])[Figure caption and citation for the preceding image starts]: Aspirado de médula ósea que muestra una célula de Gaucher típica. Se trata de un macrófago que ha ingerido material celular; el sustrato sin degradar (glucosilceramida) se acumula en los lisosomasDe la colección personal del Profesor Atul B. Mehta [Citation ends].
 [Figure caption and citation for the preceding image starts]: Resonancia magnética del esqueleto en la enfermedad de Gaucher tipo 1 que muestra una deposición generalizada de sustrato en el esqueleto con necrosis asociada e infarto óseo. Se ha producido necrosis en la cabeza del fémur (flecha)De la colección personal del Profesor Atul B. Mehta [Citation ends].
[Figure caption and citation for the preceding image starts]: Resonancia magnética del esqueleto en la enfermedad de Gaucher tipo 1 que muestra una deposición generalizada de sustrato en el esqueleto con necrosis asociada e infarto óseo. Se ha producido necrosis en la cabeza del fémur (flecha)De la colección personal del Profesor Atul B. Mehta [Citation ends].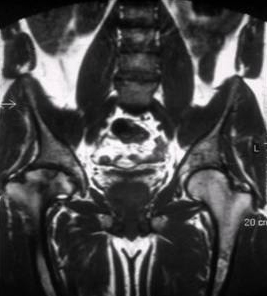
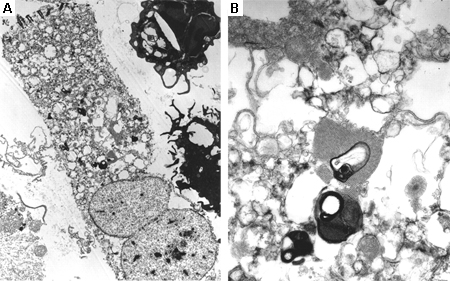
Enfermedad de Fabry (deficiencia de alfa-galactosidasa)[Figure caption and citation for the preceding image starts]: Imagen de microscopio electrónico de una biopsia de células epiteliales pulmonares en la enfermedad de Fabry que muestra los depósitos de sustrato característicos en los lisosomas, que forman "cuerpos de cebra": (A) ampliación x8000, (B) ampliación x62,500Kelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. Agosto 2000; 55 (8): 720-1; usado con autorización [Citation ends].
 [Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) palmas de las manos, (B) labios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].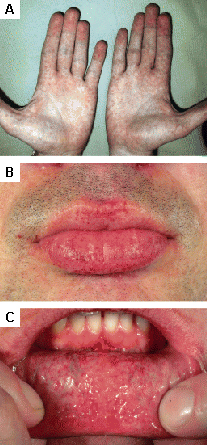 [Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) fosa lumbar, (B) genitales, (C) ombligo, (D) zona lumbar, (E) dedos de los piesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesiones cutáneas en la enfermedad de Fabry: (A) fosa lumbar, (B) genitales, (C) ombligo, (D) zona lumbar, (E) dedos de los piesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: datos de FOS, the Fabry Outcome Survey. Br J Dermatol. Agosto 2007;157(2):331-7; usado con autorización [Citation ends].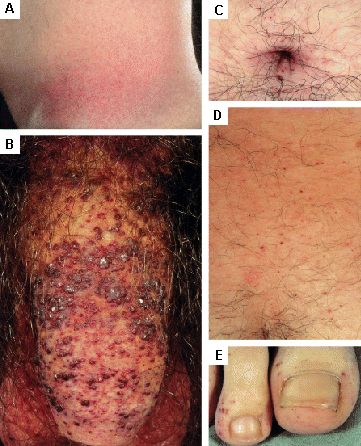
enfermedad de Tay-Sachs (deficiencia de beta-hexosaminidasa A)
enfermedad de Sandhoff (deficiencia de beta-hexosaminidasa B)
enfermedad de Krabbe (deficiencia de beta-galactosilceramidasa)
enfermedad de Niemann-Pick tipos A y B (deficiencia de esfingomielinasa ácida)
enfermedad de Niemann-Pick tipo C (defecto del transporte complejo)
leucodistrofia metacromática (deficiencia de arilsulfatasa A)
Gangliosidosis GM1 (deficiencia de beta-galactosidasa)
Mucopolisacaridosis (MPS)
MPS I (deficiencia de alfa-L-iduronidasa) en formas graves (Hurler) y formas más leves (Hurler-Scheie, Scheie)
MPS II/síndrome de Hunter (deficiencia de iduronato-2-sulfatasa)
MPS III/síndrome de Sanfilippo (4 subtipos: A, B, C, D) (deficiencia de heparán-N-sulfatasa y otras enzimas)
MPS IV/síndrome de Morquio (2 subtipos: A, B) (deficiencia de N-acetilgalactosamina 6-sulfatasa y beta-galactosidasa)
MPS VI/síndrome de Maroteaux-Lamy (deficiencia de N-acetilgalactosamina 4-sulfatasa)
MPS VII/síndrome de Sly (deficiencia de beta-glucuronidasa)
MPS IX/síndrome de Natowicz (deficiencia de hialuronidasa)
Deficiencia de MPS X/Arilsulfatasa K
Otros
Enfermedad de Pompe/enfermedad del almacenamiento del glucógeno tipo 2 (deficiencia de maltasa ácida).
glucoproteinosis (p. ej., aspartilglucosaminuria, fucosidosis, alfa y beta manosidosis, enfermedad de Schindler)
Deficiencia múltiple de sulfatasa, mucolipidosis II/III y IV, enfermedad de Danon, enfermedad de Wolman, defectos del transporte lisosomal (p. ej., cistinosis, enfermedad de almacenamiento de ácido siálico)
Fisiopatología
La deficiencia enzimática conduce a un metabolismo intermediario alterado con acumulación de sustrato no degradado.[1][2] Dicha acumulación no solo conduce a un agrandamiento de las células y órganos individuales, sino que también causa anomalías secundarias bioquímicas y estructurales. Se produce hipertrofia lisosómica, que resulta en una alteración macroscópica de la función lisosómica.
Los cambios neuronales son parcialmente estructurales y se deben al aumento del almacenamiento; no obstante, la apoptosis neuronal acelerada ha quedado demostrada en las mucopolisacaridosis y el metabolismo alterado del calcio que deriva en una muerte celular neuronal excesiva ha sido demostrado en el caso de la enfermedad de Gaucher neuronopática.[24][25]
La acumulación extralisosómica de sustrato puede alterar la función de la membrana, el metabolismo mitocondrial, los canales y transportadores iónicos de la membrana y la transducción de señales a través de las "balsas lipídicas" de la membrana.[1][26]
La activación de los macrófagos, posiblemente mediada por la liberación de citoquinas, se ha demostrado en la enfermedad de Gaucher y en otras enfermedades de almacenamiento lisosomal (EDL).[27][28] La acumulación de lisoesfingolípidos (es decir, glucoesfingolípidos sin ácidos grasos N-acilados) puede ser de importancia patogénica en las enfermedades de Krabbe, Gaucher y Fabry.[29]
Fisiopatología específica de la enfermedad
Enfermedad de Gaucher: por deficiencia de beta-glucocerebrosidasa. Esto hace que el lisosoma sea incapaz de descomponer los glicoesfingolípidos, lo que conduce a una acumulación de glucosilceramidas.[30]
Enfermedad de Fabry: causada por la deficiencia de alfa-galactosidasa, que provoca la acumulación de globotriaosilceramida en los lisosomas.[31]
Enfermedad de Tay-Sachs: resultados de la deficiencia de beta-hexosaminidasa A. Los gangliósidos se acumulan en los lisosomas, especialmente en las neuronas.[32]
Los tipos A y B de la enfermedad de Niemann-Pick (ENP) se deben a la deficiencia de esfingomielinasa. La ENP tipo A es un trastorno neurodegenerativo grave que se suele presentar en la lactancia, mientras que la ENP tipo B, que generalmente no afecta el sistema nervioso, se caracteriza por la acumulación de sustrato en el sistema reticuloendotelial (causa hepatoesplenomegalia, enfermedad pulmonar) y presenta más tarde. La ENP tipo C es un trastorno complejo del transporte y procesamiento intracelular del colesterol, que generalmente se presenta en la infancia con ataxia, trastorno del movimiento ocular y dificultades de aprendizaje; no obstante, puede darse en adultos como una enfermedad neurológica o con hepatoesplenomegalia.[33]
Mucopolisacaridosis: se caracterizan por una descomposición incompleta y una acumulación progresiva de glicosaminoglicanos.[34]
Enfermedad de Pompe: causada por la deficiencia de maltasa ácida, que conduce a la acumulación de glucógeno en los lisosomas.[35]
Clasificación
Esfingolipidosis
Incidencia: >1/50,000 personas[5]
Enfermedad de Gaucher (deficiencia de beta-glucocerebrosidasa) tipo 1 (la forma más frecuente), tipos 2 y 3 (con afectación del sistema nervioso central)
Enfermedad de Fabry (deficiencia de alfa-galactosidasa)
Incidencia: <1/100,000 personas[5]
enfermedad de Tay-Sachs (deficiencia de beta-hexosaminidasa A)
enfermedad de Sandhoff (deficiencia de beta-hexosaminidasa B)
enfermedad de Krabbe (deficiencia de beta-galactosilceramidasa)
enfermedad de Niemann-Pick tipos A y B (deficiencia de esfingomielinasa ácida)
enfermedad de Niemann-Pick tipo C (defecto del transporte complejo)
leucodistrofia metacromática (deficiencia de arilsulfatasa A)
Gangliosidosis GM1 (deficiencia de beta-galactosidasa)
Mucopolisacaridosis (MPS)
Incidencia: >1/100,000 personas[5]
MPS I (deficiencia de alfa-L-iduronidasa) en formas graves (Hurler) y formas más leves (Hurler-Scheie, Scheie)
MPS II/síndrome de Hunter (deficiencia de iduronato-2-sulfatasa)
Incidencia: <1/100,000 personas[5]
MPS III/síndrome de Sanfilippo (4 subtipos: A, B, C, D) (deficiencia de heparán-N-sulfatasa y otras enzimas)
MPS IV/síndrome de Morquio (2 subtipos: A, B) (deficiencia de N-acetilgalactosamina 6-sulfatasa y beta-galactosidasa)
MPS VI/síndrome de Maroteaux-Lamy (deficiencia de N-acetilgalactosamina 4-sulfatasa)
MPS VII/síndrome de Sly (deficiencia de beta-glucuronidasa)
MPS IX/síndrome de Natowicz (deficiencia de hialuronidasa)
Deficiencia de MPS X/Arilsulfatasa K
Otros
Incidencia: >1/50,000 personas[5]
Enfermedad de Pompe/enfermedad del almacenamiento del glucógeno tipo 2 (deficiencia de maltasa ácida)
Incidencia: >1/100,000 personas[5]
glucoproteinosis (p. ej., aspartilglucosaminuria, fucosidosis, alfa y beta manosidosis, enfermedad de Schindler)
Deficiencia múltiple de sulfatasa, mucolipidosis II/III y IV, enfermedad de Danon, enfermedad de Wolman, defectos del transporte lisosomal (p. ej., cistinosis, enfermedad de almacenamiento de ácido siálico)
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad