As maiores manifestações de deficiência de alfa 1-antitripsina (AAT) são hepáticas e pulmonares.
As manifestações mais comuns são o enfisema panacinar e a doença pulmonar obstrutiva associada.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[38]Marciniuk DD, Hernandez P, Balter M, et al. Alpha-1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clinical practice guideline. Can Respir J. 2012;19:109-116.
https://www.hindawi.com/journals/crj/2012/920918
http://www.ncbi.nlm.nih.gov/pubmed/22536580?tool=bestpractice.com
Evidências sugerem que quase 60% dos pacientes desenvolvem doença pulmonar grave.[23]Larsson C. Natural history and life expectancy in severe alpha 1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204:345-351.
http://www.ncbi.nlm.nih.gov/pubmed/309708?tool=bestpractice.com
Podem ocorrer bronquiectasias. Até 95% dos pacientes com deficiência de AAT de PI*ZZ apresentam evidências radiológicas de bronquiectasia (mas a deficiência de AAT de PI*ZZ foi encontrada em <1% dos pacientes com bronquiectasia).[39]Hill AT, Sullivan AL, Chalmers JD, et al. British Thoracic Society Guideline for bronchiectasis in adults. Thorax. 2019 Jan;74(suppl 1):1-69.
https://thorax.bmj.com/content/74/Suppl_1/1.long
http://www.ncbi.nlm.nih.gov/pubmed/30545985?tool=bestpractice.com
A doença hepática costuma apresentar-se, inicialmente, como hepatite e icterícia, embora a doença grave possa evoluir para cirrose e carcinoma hepatocelular. O envolvimento hepático também pode ser observado em neonatos com deficiência de AAT.[8]Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018 Dec;24 Suppl 1:1-21.
https://www.doi.org/10.1016/j.pulmoe.2018.09.004
http://www.ncbi.nlm.nih.gov/pubmed/30473034?tool=bestpractice.com
[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
A granulomatose com poliangiite e a paniculite necrosante são complicações pouco comuns.
As evidências sugerem que a deficiência de AAT pode ser pouco reconhecida por médicos como uma causa de doença pulmonar e hepática.[40]Silverman EK, Miletich JP, Pierce JA, et al. Alpha-1 antitrypsin deficiency: high prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis. 1989;140:961-966.
http://www.ncbi.nlm.nih.gov/pubmed/2679271?tool=bestpractice.com
Outras evidências demonstram uma diferença de tempo significativa entre o início da doença clínica e o diagnóstico, com avaliação por vários médicos nesse meio tempo.[41]Stoller JK, Smith P, Yang P, et al. Physical and social impact of alpha-1 antitrypsin deficiency: results of a survey. Cleve Clin J Med. 1994;61:461-467.
http://www.ncbi.nlm.nih.gov/pubmed/7828337?tool=bestpractice.com
As diretrizes recomendam uma alta suspeita clínica e a medição quantitativa de AAT nos seguintes cenários:[5]American Thoracic Society/European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha 1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003 Oct 1;168(7):818-900.
https://www.atsjournals.org/doi/full/10.1164/rccm.168.7.818
http://www.ncbi.nlm.nih.gov/pubmed/14522813?tool=bestpractice.com
[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[42]World Health Organization. Alpha-1 antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75:397-415.
http://www.ncbi.nlm.nih.gov/pubmed/9447774?tool=bestpractice.com
[43]Eriksson S, Carlson J, Velez R. Risks for cirrhosis and primary liver cancer in alpha-1 antitrypsin deficiency. N Engl J Med. 1986;314:736-739.
http://www.ncbi.nlm.nih.gov/pubmed/3485248?tool=bestpractice.com
Obstrução do fluxo aéreo parcialmente reversível ou irreversível com broncodilatadores
Todos os pacientes com DPOC[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[44]Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for prevention, diagnosis and management of COPD: 2024 report. 2024 [internet publication].
https://goldcopd.org/2024-gold-report
Todos os pacientes com asma de início na fase adulta[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
História familiar ou pessoal de vasculite associada a anticorpos anticitoplasma de neutrófilos (c-ANCA) (a granulomatose com poliangiite é uma complicação pouco frequente da deficiência de AAT)
Doença hepática de etiologia desconhecida
Bronquiectasia de etiologia desconhecida, principalmente quando coexistente com enfisema panacinar[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[39]Hill AT, Sullivan AL, Chalmers JD, et al. British Thoracic Society Guideline for bronchiectasis in adults. Thorax. 2019 Jan;74(suppl 1):1-69.
https://thorax.bmj.com/content/74/Suppl_1/1.long
http://www.ncbi.nlm.nih.gov/pubmed/30545985?tool=bestpractice.com
Paniculite de etiologia desconhecida (a paniculite necrosante é uma complicação pouco frequente da deficiência de AAT)
Adolescentes e adultos com um irmão que seja homozigoto para AAT
Pessoas assintomáticas com disfunção pulmonar obstrutiva persistente que relatam tabagismo ou exposição ocupacional.
Fatores históricos
Os sintomas manifestos de doenças pulmonares são inespecíficos e podem incluir dispneia, dispneia durante o esforço físico, fadiga, sibilância, tosse e/ou constrição torácica.
Os sintomas manifestos de doenças hepáticas são inespecíficos e podem incluir amarelamento da pele, fadiga, sangramento, hematomas, distensão abdominal, dor abdominal e/ou confusão.
É importante considerar idade, ocupação e história de tabagismo em pacientes com doença pulmonar sintomática, pois esses fatores podem apontar para uma deficiência de AAT. O maior fator de risco para enfisema em pacientes com o fenótipo PI*ZZ é o tabagismo. Tanto a função pulmonar quanto a sobrevida são afetadas.[23]Larsson C. Natural history and life expectancy in severe alpha 1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204:345-351.
http://www.ncbi.nlm.nih.gov/pubmed/309708?tool=bestpractice.com
[45]Janus ED, Phillips NT, Carrell RW. Smoking, lung function, and alpha-1-antitrypsin deficiency. Lancet. 1985;1:152-4.
http://www.ncbi.nlm.nih.gov/pubmed/2857224?tool=bestpractice.com
[46]Wu MC, Eriksson S. Lung function, smoking and survival in severe alpha 1-antitrypsin deficiency, PiZZ. J Clin Epidemiol. 1988;41:1157-1165.
http://www.ncbi.nlm.nih.gov/pubmed/3264848?tool=bestpractice.com
No entanto, certas evidências sugerem que ex-fumantes e pessoas que nunca fumaram têm declínios similares na função pulmonar ao longo do tempo, e alguns fumantes podem nunca desenvolver sintomas pulmonares.[41]Stoller JK, Smith P, Yang P, et al. Physical and social impact of alpha-1 antitrypsin deficiency: results of a survey. Cleve Clin J Med. 1994;61:461-467.
http://www.ncbi.nlm.nih.gov/pubmed/7828337?tool=bestpractice.com
[47]Piitulainen E, Eriksson S. Decline in FEV1 related to smoking status in individuals with severe alpha-1 antitrypsin deficiency (PiZZ). Eur Respir J. 1999;13:247-251.
http://erj.ersjournals.com/cgi/reprint/13/2/247
http://www.ncbi.nlm.nih.gov/pubmed/10065663?tool=bestpractice.com
A exposição a gases, vapores e/ou poeira, seja ocupacional ou por outros meios, também foi associada à função pulmonar reduzida em pacientes com deficiência de AAT com fenótipo PI*ZZ. Isso inclui tabagismo passivo e trabalho com aquecedores a querosene.[48]Piitulainen E, Tornling G, Eriksson S. Effect of age and occupational exposure to airway irritants on lung function in non-smoking individuals with alpha-1 antitrypsin deficiency (PiZZ). Thorax. 1997;52:244-248.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1758519/pdf/v052p00244.pdf
http://www.ncbi.nlm.nih.gov/pubmed/9093340?tool=bestpractice.com
[49]Piitulainen E, Tornling G, Eriksson S. Environmental correlates of impaired lung function in non-smokers with severe alpha-1 antitrypsin deficiency (PiZZ). Thorax. 1998;53:939-943.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1745103/pdf/v053p00939.pdf
http://www.ncbi.nlm.nih.gov/pubmed/10193391?tool=bestpractice.com
[50]Silverman EK, Pierce JA, Province MA, et al. Variability of pulmonary function in alpha-1 antitrypsin deficiency: clinical correlates. Ann Intern Med. 1989;111:982-991.
http://www.ncbi.nlm.nih.gov/pubmed/2596778?tool=bestpractice.com
[51]Piitulainen E, Sveger T. Effect of environmental and clinical factors on lung function and respiratory symptoms in adolescents with alpha-1 antitrypsin deficiency. Acta Paediatr. 1998;87:1120-1124.
http://www.ncbi.nlm.nih.gov/pubmed/9846912?tool=bestpractice.com
Evidências limitadas sugerem que a doença pulmonar sintomática seja mais prevalente em homens PI*ZZ que em mulheres PI*ZZ. No entanto, é provável que esse resultado seja mascarado por outras variáveis, como tabagismo e exposição ocupacional.[19]Kueppers F, Fallat R, Larson RK. Obstructive lung disease and alpha-1 antitrypsin deficiency gene heterozygosity. Science. 1969;165:899-901.
http://www.ncbi.nlm.nih.gov/pubmed/5816326?tool=bestpractice.com
[20]Kueppers F, Black LF. Alpha-1 antitrypsin and its deficiency. Am Rev Respir Dis. 1974;110:176-194.
http://www.ncbi.nlm.nih.gov/pubmed/4212922?tool=bestpractice.com
[21]Tobin MJ, Cook PJ, Hutchison DC. Alpha-1 antitrypsin deficiency: the clinical and physiological features of pulmonary emphysema in subjects homozygous for Pi-type-Z. A survey by the British Thoracic Association. Br J Dis Chest. 1983;77:14-27.
http://www.ncbi.nlm.nih.gov/pubmed/6602621?tool=bestpractice.com
[22]Seersholm N, Kok-Jensen A, Dirksen A. Decline in FEV1 among patients with severe hereditary alpha-1 antitrypsin deficiency type Pi Z. Am J Respir Crit Care Med. 1995;152:1922-1925.
http://www.ncbi.nlm.nih.gov/pubmed/8520756?tool=bestpractice.com
A idade média de manifestação de doença pulmonar sintomática em fumantes com deficiência de AAT é tipicamente entre 32 e 41 anos.[23]Larsson C. Natural history and life expectancy in severe alpha 1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204:345-351.
http://www.ncbi.nlm.nih.gov/pubmed/309708?tool=bestpractice.com
A história médica pode incluir asma e/ou granulomatose com poliangiite (uma complicação pouco frequente da deficiência de AAT), e a história familiar pode revelar deficiência de AAT em parentes.
Achados dos exames
Uma inspeção geral pode revelar icterícia, esclerótica ictérica e/ou asterixis (flapping) em caso de doença hepática estar presente. Um exame abdominal pode revelar hepatomegalia e/ou ascite.
Um exame respiratório pode revelar sibilo e/ou hiperinsuflação torácica em caso de doença pulmonar.
Medições da AAT sérica
Os níveis séricos de AAT devem ser quantificados em indivíduos com possível deficiência de AAT.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
No entanto, a AAT também é um reagente de fase aguda, o que significa que os níveis normais de AAT sérica podem ser enganosos, especialmente em cenários de processos inflamatórios.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
Estados patológicos podem ser representados, ainda, por níveis limítrofes ou mesmo normais de AAT, o que significa que tais resultados justifiquem suspeita contínua. A medição sérica de AAT isolada não é recomendada para testes familiares após a identificação de um probando porque não caracteriza totalmente o risco de doença decorrente da deficiência de AAT.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
Algumas diretrizes sugerem a genotipagem para os alelos S e Z como sendo a primeira etapa adequada para testes diagnósticos de indivíduos sintomáticos.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
Testes quantitativos e limiar de proteção
Níveis baixos a normais de AAT (<35 micromoles/L) devem aumentar a suspeita e incitar testes imediatos adicionais. Testes quantitativos comercialmente disponíveis utilizam imunodifusão radial e métodos de nefelometria. O valor exato do limite varia de acordo com o método de teste e a orientação regional; as diretrizes regionais adequadas devem ser consultadas para interpretação dos níveis séricos de AAT.[5]American Thoracic Society/European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha 1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003 Oct 1;168(7):818-900.
https://www.atsjournals.org/doi/full/10.1164/rccm.168.7.818
http://www.ncbi.nlm.nih.gov/pubmed/14522813?tool=bestpractice.com
[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[8]Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018 Dec;24 Suppl 1:1-21.
https://www.doi.org/10.1016/j.pulmoe.2018.09.004
http://www.ncbi.nlm.nih.gov/pubmed/30473034?tool=bestpractice.com
[9]Dummer J, Dobler CC, Holmes M, et al. Diagnosis and treatment of lung disease associated with alpha one-antitrypsin deficiency: a position statement from the Thoracic Society of Australia and New Zealand. Respirology. 2020 Mar;25(3):321-35.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7078913
http://www.ncbi.nlm.nih.gov/pubmed/32030868?tool=bestpractice.com
O nível sérico de AAT medido usando o padrão purificado desenvolvido pelo National Institutes of Health dos EUA (o método de teste mais comum nos EUA) geralmente é representado em micromoles/L, enquanto os níveis de AAT medidos usando os padrões comerciais geralmente são representados em mg/dL para diferenciá-los.[5]American Thoracic Society/European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha 1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003 Oct 1;168(7):818-900.
https://www.atsjournals.org/doi/full/10.1164/rccm.168.7.818
http://www.ncbi.nlm.nih.gov/pubmed/14522813?tool=bestpractice.com
Valores de nefelometria menores que 20 micromoles/L (83-120 mg/dL) são considerados deficientes. Considera-se que níveis de nefelometria abaixo de 11 micromoles/L (50 mg/dL) ofereçam proteção inadequada contra doenças pulmonares inflamatórias; isso é chamado de "limiar de proteção".[6]Turino GM, Barker AF, Brantly ML, et al. Clinical features of individuals with PI*SZ phenotype of alpha-1 antitrypsin deficiency: alpha 1-antitrypsin deficiency registry study group. Am J Respir Crit Care Med. 1996;154:1718-1725.
http://www.ncbi.nlm.nih.gov/pubmed/8970361?tool=bestpractice.com
Alguns dos fenótipos mais comuns resultam nos seguinte níveis de AAT sérica:
PI*MM: 20-48 micromoles/L (150-350 mg/dL)
PI*MZ: 17-33 micromoles/L (90-210 mg/dL)
PI*SS: 15-33 micromoles/L (100-200 mg/dL)
PI*ZZ: 2.5 -7.0 micromoles/L (20-45 mg/dL).
Os fenótipos que resultam em níveis abaixo do limiar de proteção têm maior probabilidade de resultar em doença pulmonar.
Fenotipagem (tipagem PI)
Medições de AAT plasmática baixas a normais podem corresponder a fenótipos heterozigotos que podem colocar o indivíduo e os familiares em risco para doenças associadas. Pacientes e seus familiares de primeiro grau com níveis de AAT normais a baixos porém protetores (12-35 micromoles/L) devem submeter-se a exames qualitativos por fenotipagem.
A fenotipagem envolve a separação de variantes de AAT utilizando focalização isoelétrica e pode confirmar a identificação de proteínas variantes de AAT deficientes características. A fenotipagem pode revelar a real presença das variantes de proteínas, como a proteína Z, proteína M (normal) e proteína S, bem como variantes menos comuns.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
Genotipagem
O teste genético pode ser realizado quando o fenótipo real não corresponde ao fenótipo predito pelo nível de AAT sérica.
Ele demonstrará os alelos de AAT característicos responsáveis pelas proteínas variantes de AAT.
Por exemplo, quando um nível de AAT baixo a normal é detectado, realizam-se testes adicionais com fenotipagem a fim de determinar as variantes reais de proteína AAT no soro. Se for detectada apenas a proteína Z, isso não corresponde a um nível de AAT sérica de baixo a normal. Nesse caso, podem-se realizar testes adicionais de genotipagem para determinar os alelos presentes no indivíduo.
Geralmente, a reação em cadeia da polimerase é usada para genotipagem. Alelos raros (por exemplo, variantes nulas ou deficientes diferentes de Z ou S) podem requerer um sequenciamento do gene completo. O sequenciamento do gene também pode ser considerado caso não haja primers disponíveis para a reação em cadeia da polimerase.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[9]Dummer J, Dobler CC, Holmes M, et al. Diagnosis and treatment of lung disease associated with alpha one-antitrypsin deficiency: a position statement from the Thoracic Society of Australia and New Zealand. Respirology. 2020 Mar;25(3):321-35.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7078913
http://www.ncbi.nlm.nih.gov/pubmed/32030868?tool=bestpractice.com
Exames específicos para doença respiratória
Se houver doença respiratória, os testes da função pulmonar mostrarão resultados significativamente anormais, incluindo VEF1 reduzido.
Uma radiografia torácica pode revelar grandes volumes pulmonares e enfisema predominante basilar.[Figure caption and citation for the preceding image starts]: Radiografia torácica de deficiência de alfa 1-antitripsina (AAT) (visualização póstero-anterior [PA])Da coleção pessoal de D. Kyle Hogarth, MD, FCCP; usado com permissão [Citation ends].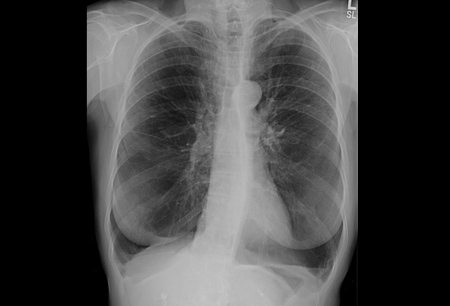 [Figure caption and citation for the preceding image starts]: Radiografia torácica de deficiência de alfa 1-antitripsina (AAT) (visualização lateral)Da coleção pessoal de D. Kyle Hogarth, MD, FCCP; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Radiografia torácica de deficiência de alfa 1-antitripsina (AAT) (visualização lateral)Da coleção pessoal de D. Kyle Hogarth, MD, FCCP; usado com permissão [Citation ends].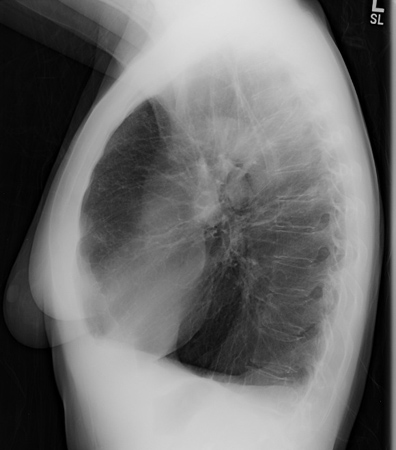
Pacientes com resultados não diagnósticos podem necessitar de uma tomografia computadorizada (TC) do tórax. A TC é mais sensível que a radiografia torácica ou que os testes da função pulmonar, pois ela identifica o enfisema panacinar e as bronquiectasias.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[39]Hill AT, Sullivan AL, Chalmers JD, et al. British Thoracic Society Guideline for bronchiectasis in adults. Thorax. 2019 Jan;74(suppl 1):1-69.
https://thorax.bmj.com/content/74/Suppl_1/1.long
http://www.ncbi.nlm.nih.gov/pubmed/30545985?tool=bestpractice.com
No entanto, a ausência de alterações enfisematosas na TC não descarta a deficiência de AAT. O enfisema panacinar é predominantemente observado nos lobos inferiores, embora a doença também tenha sido descrita no lobo superior somente. Uma relação direta entre deficiência de AAT e bronquiectasia é menos evidente, pois a presença de bronquiectasia na TC pode ser resultado das alterações enfisematosas.[Figure caption and citation for the preceding image starts]: TC de enfisema avançado em paciente com deficiência de AATDa coleção pessoal de D. Kyle Hogarth, MD, FCCP; usado com permissão [Citation ends].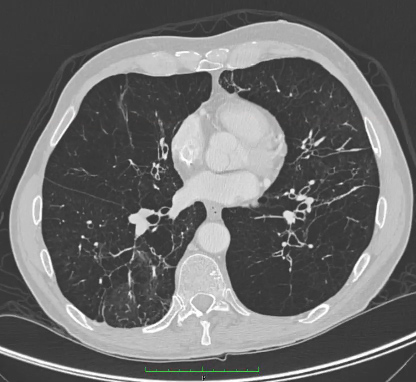
Testes ergométricos com análise de gasometria arterial em pacientes com enfisema também costumam ser anormais e demonstram intolerância ao exercício.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[8]Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018 Dec;24 Suppl 1:1-21.
https://www.doi.org/10.1016/j.pulmoe.2018.09.004
http://www.ncbi.nlm.nih.gov/pubmed/30473034?tool=bestpractice.com
Exames específicos para doença hepática
As diretrizes recomendam avaliação da função hepática com testes da função hepática (TFHs) para pacientes diagnosticados com deficiência de AAT, quer sejam sintomáticos ou assintomáticos para doença hepática.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
Os níveis de alfafetoproteína (AFP) também são importantes como parte de qualquer investigação de doença hepática. Porém, alguns dados sugerem que a sensibilidade dos testes da função hepática, mais precisamente da alanina aminotransferase (ALT), é de apenas 11.9% na detecção de doença hepática na deficiência de AAT.[53]Clark VC, Dhanasekaran R, Brantly M, et al. Liver test results do not identify liver disease in adults with alpha-1 antitrypsin deficiency. Clin Gastroenterol Hepatol. 2012;10:1278-1283.
http://www.ncbi.nlm.nih.gov/pubmed/22835581?tool=bestpractice.com
Os pacientes com fenótipo associado à doença hepática (por exemplo, PI*ZZ, PI*Mmalton, PI*Siiyama) requerem exames de imagem do fígado, e a ultrassonografia do fígado é recomendada anualmente.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
A ultrassonografia do fígado também pode ser usada para monitorar sinais de hipertensão portal e carcinoma hepatocelular. A TC abdominal e/ou a ressonância nuclear magnética (RNM) também podem ser úteis para avaliar os pacientes em relação a morfologia do fígado, cirrose e hipertensão portal, particularmente naqueles com obesidade.[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
Se houver carcinoma hepatocelular, os testes da função hepática (TFHs) podem apresentar resultados cada vez piores, e os níveis de AFP podem aumentar.