As doenças do armazenamento lisossomal (DALs) são um grupo altamente diversificado de doenças. O tratamento inicial é geral e de suporte. Uma abordagem multidisciplinar é essencial, pois essas doenças são distúrbios multissistêmicos; muitos especialistas diferentes podem ser envolvidos no cuidado dos pacientes individuais. É recomendável o envolvimento precoce de centros especializados, e os cuidados devem ser coordenados pelo centro. O tratamento específico não está disponível para muitos pacientes (por exemplo, doença de Gaucher neonatal do tipo 2), e a paliação pode ser o melhor caminho.
A terapia de reposição enzimática (TRE) foi estabelecida para várias DALs.[3]Mehta A, Winchester B, eds. Lysosomal storage disorders: a practical guide. 2nd ed. Oxford: Wiley-Blackwell; 2022. Na Europa, a TRE pode ser administrada na própria casa do paciente para distúrbios de Gaucher, Fabry, Pompe, mucopolissacaridose (MPS) I e MPS II.
Fisioterapeutas, terapeutas ocupacionais, especialistas em cuidados clínicos, psicólogos, professores e assistentes sociais são todos membros apropriados da equipe multidisciplinar ampliada.
Doenças de Gaucher
Doença de Gaucher do tipo 1
O atraso no diagnóstico continua sendo uma preocupação, e há uma necessidade de aumentar a conscientização entre médicos generalistas, pediatras e hematologistas.[98]Kishnani PS, Al-Hertani W, Balwani M, et al. Screening, patient identification, evaluation, and treatment in patients with Gaucher disease: results from a Delphi consensus. Mol Genet Metab. 2022 Feb;135(2):154-62.
https://www.sciencedirect.com/science/article/pii/S109671922101194X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/34972655?tool=bestpractice.com
Pessoas levemente afetadas, por exemplo, com hemoglobina normal, plaquetas >100 x 10⁹/L, aumento mínimo do fígado e baço, sem lesões ósseas na ressonância nuclear magnética (RNM), justificam observação, mas podem não precisar de tratamento específico. Pacientes assintomáticos não exigem tratamento.[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58.
http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com
A esplenectomia deve ser evitada; ela melhora as contagens sanguíneas, mas predispõe à doença óssea mais grave em longo prazo e à hipertensão pulmonar.[100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86.
https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x
http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com
Pode haver doença esquelética que causa dor óssea ou fratura; pode ser necessária cirurgia ortopédica para tratar as articulações afetadas.
O aumento da incidência de neoplasias hematológicas em adultos deve ser monitorado quanto a: especialmente mieloma, mas também outras doenças, como linfoma não Hodgkin.[100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86.
https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x
http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com
Doença de Parkinson e cirrose hepática são outras complicações em longo prazo que requerem monitoramento e tratamento.
Anormalidades endócrinas e metabólicas coexistentes, incluindo resistência à insulina, deficiência de hormônio do crescimento e insuficiência de vitamina D, devem ser investigadas e tratadas.[101]Kałużna M, Trzeciak I, Ziemnicka K, et al. Endocrine and metabolic disorders in patients with Gaucher disease type 1: a review. Orphanet J Rare Dis. 2019 Dec 2;14(1):275.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-019-1211-5
http://www.ncbi.nlm.nih.gov/pubmed/31791361?tool=bestpractice.com
TRE com imiglucerase, velaglucerase alfa e taliglucerase alfa está disponível.[102]Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010 Jun 10;115(23):4651-6.
http://www.bloodjournal.org/content/115/23/4651.long
http://www.ncbi.nlm.nih.gov/pubmed/20299511?tool=bestpractice.com
[103]Morris JL. Velaglucerase alfa for the management of type 1 Gaucher disease. Clin Ther. 2012 Feb;34(2):259-71.
http://www.ncbi.nlm.nih.gov/pubmed/22264444?tool=bestpractice.com
[104]Ben Turkia H, Gonzalez DE, Barton NW, et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am J Hematol. 2013 Mar;88(3):179-84.
http://www.ncbi.nlm.nih.gov/pubmed/23400823?tool=bestpractice.com
[105]Hughes DA, Gonzalez DE, Lukina EA, et al. Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: long-term data from phase III clinical trials. Am J Hematol. 2015 Jul;90(7):584-91.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4654249
http://www.ncbi.nlm.nih.gov/pubmed/25801797?tool=bestpractice.com
A TRE deve ser considerada em todas as crianças sintomáticas com doença de Gaucher, e nos adultos com reduções significativas no hemograma (por exemplo, nível de hemoglobina <100 g/L [10 g/dL], plaquetas <100 x 10⁹/L), aumento significativo de órgãos (por exemplo, tamanho do baço >10x o normal), presença de doença esquelética demonstrada na RNM e/ou qualquer outro dano a órgãos (por exemplo, evidência de lesão no pulmão).[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58.
http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com
[100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86.
https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x
http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com
A TRE tem benefício comprovado na melhora de anormalidades hematológicas e dores ósseas, e na redução do tamanho do fígado e baço. Densidade óssea, função pulmonar e qualidade de vida também melhoram.[106]Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency - macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991 May 23;324(21):1464-70.
http://www.ncbi.nlm.nih.gov/pubmed/2023606?tool=bestpractice.com
[107]Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type I Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002 Aug 1;113(2):112-9.
http://www.ncbi.nlm.nih.gov/pubmed/12133749?tool=bestpractice.com
[108]Gabrowski GA, Kolodny EH, Weinreb NJ, et al. Gaucher disease: phenotypic and genetic variation. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The metabolic and molecular basis of inherited disease. 9th ed. New York, NY: McGraw-Hill; 2006.
Foi demonstrado que a terapia de redução de substrato (TRS) melhora a anemia, a trombocitopenia, o aumento do fígado/baço e a osteoporose.[109]Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015 Feb 17;313(7):695-706.
http://jama.jamanetwork.com/article.aspx?articleid=2110969
http://www.ncbi.nlm.nih.gov/pubmed/25688781?tool=bestpractice.com
A TRS (por exemplo, miglustate, eleglustate) é uma terapia oral usada em pacientes que não toleram a TRE, ou que têm acesso venoso difícil.[109]Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015 Feb 17;313(7):695-706.
http://jama.jamanetwork.com/article.aspx?articleid=2110969
http://www.ncbi.nlm.nih.gov/pubmed/25688781?tool=bestpractice.com
[110]Cox TM, Aerts JM, Andria G, et al. The role of the iminosugar N-butyldeoxynorjirimycin (miglustat) in the management of type 1 (non-neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis. 2003;26(6):513-26.
http://www.ncbi.nlm.nih.gov/pubmed/14605497?tool=bestpractice.com
[111]Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000 Apr 29;355(9214):1481-5.
http://www.ncbi.nlm.nih.gov/pubmed/10801168?tool=bestpractice.com
[112]Pastores GM, Giraldo P, Cherin P, et al. Goal-oriented therapy with miglustat in Gaucher disease. Curr Med Res Opin. 2009 Jan;25(1):23-37.
http://www.ncbi.nlm.nih.gov/pubmed/19210136?tool=bestpractice.com
[113]Poole RM. Eliglustat: first global approval. Drugs. 2014 Oct;74(15):1829-36.
http://www.ncbi.nlm.nih.gov/pubmed/25239269?tool=bestpractice.com
Os objetivos da terapia foram reconhecidos para a doença de Gaucher do tipo 1 e os tratamentos atualmente disponíveis permitem que a maioria deles seja alcançada.[114]Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: an expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis. 2018 Feb;68:203-8.
https://www.sciencedirect.com/science/article/pii/S1079979616301917?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/28274788?tool=bestpractice.com
Transplantes de células-tronco foram realizados no passado, mas a TRE é mais segura.[115]Peters C, Steward CG, National Marrow Donor Program, et al. Hematopoietic cell transplantation for inherited metabolic disorders: an overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003 Feb;31(4):229-39.
http://www.ncbi.nlm.nih.gov/pubmed/12621457?tool=bestpractice.com
Doença de Gaucher do tipo 2
A doença de Gaucher neuronopática aguda do tipo 2 deve ser tratada com medidas de suporte somente, pois a morte prematura é comum.
Complicações da doença incluem convulsões, atraso no neurodesenvolvimento e distúrbios da motilidade ocular, as quais precisam ser consideradas como parte do manejo da doença.
A doença de Gaucher do tipo 2 é essencialmente intratável.
A terapia de reposição enzimática não é efetiva.
Doença de Gaucher do tipo 3
Distúrbios da motilidade ocular, atraso no neurodesenvolvimento e doença esquelética são complicações dessa doença.
Tipicamente, acomete crianças e adultos jovens; os aspectos viscerais e esqueléticos da doença respondem bem à TRE, mas não há melhora das manifestações neurológicas, pois a TRE não consegue atravessar a barreira hematoencefálica.[116]Vellodi A, Tylki-Szymanska A, Davies EH, et al. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis. 2009 Oct;32(5):660-4.
http://www.ncbi.nlm.nih.gov/pubmed/19655269?tool=bestpractice.com
Doença de Fabry
Há um grande número de aspectos gerais de suporte no manejo dessa doença multissistêmica.
O alívio da dor com gabapentina ou pregabalina é indicado para dor neuropática. Carbamazepina também é amplamente usada.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
[117]Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018 Jul;124(3):189-203.
http://www.ncbi.nlm.nih.gov/pubmed/30017653?tool=bestpractice.com
Anti-inflamatórios não esteroidais devem ser usados com moderação, pois esses pacientes muitas vezes apresentam nefropatia.
As lesões cutâneas podem necessitar de cirurgia estética ou laserterapia.[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) flanco, (B) genitais, (C) umbigo, (D) coluna lombar, (E) pododáctilosOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].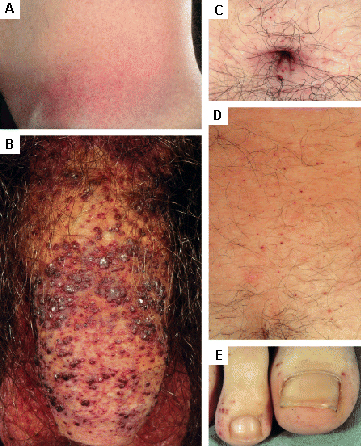 [Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].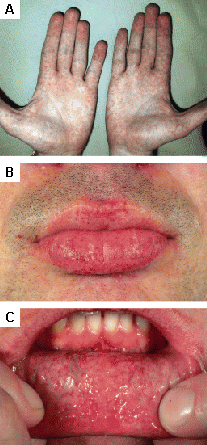
A pressão arterial e a proteinúria devem ser monitoradas regularmente com intervenção precoce de inibidores da enzima conversora da angiotensina (ECA) ou antagonistas do receptor de angiotensina II.[118]Jain G, Warnock DG. Blood pressure, proteinuria and nephropathy in Fabry disease. Nephron Clin Pract. 2011;118(1):43-8.
http://www.ncbi.nlm.nih.gov/pubmed/21071972?tool=bestpractice.com
A avaliação cardíaca é essencial; o reconhecimento precoce de arritmia é importante. Cirurgia, incluindo inserção de marca-passos, ressecção do septo, substituição da valva, e até mesmo o transplante cardíaco, deve ser considerada.[119]Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage diseases. Heart. 2007 Apr;93(4):528-35.
http://www.ncbi.nlm.nih.gov/pubmed/17401074?tool=bestpractice.com
O AVC e o ataque isquêmico transitório requerem cuidadosas medidas preventivas primárias e secundárias.
Avaliação gastrointestinal, terapia sintomática e endoscopia podem ser necessárias para descartar as condições associadas.[120]Hoffmann B, Schwarz M, Mehta A, et al. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007 Dec;5(12):1447-53.
http://www.ncbi.nlm.nih.gov/pubmed/17919989?tool=bestpractice.com
A depressão é comum na doença de Fabry; a família deve ser observada em conjunto, sempre que possível. Aconselhamento pode ser necessário.
Surdez é altamente provável.[121]Germain DP, Avan P, Chassaing A, et al. Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty-two hemizygous male patients. BMC Med Genet. 2002 Oct 11;3:10.
https://bmcmedgenet.biomedcentral.com/articles/10.1186/1471-2350-3-10
http://www.ncbi.nlm.nih.gov/pubmed/12377100?tool=bestpractice.com
Tratamento específico com TRE ou terapia com chaperonas
Diretrizes para diagnóstico e tratamento estão disponíveis e são atualizadas regularmente.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
[117]Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018 Jul;124(3):189-203.
http://www.ncbi.nlm.nih.gov/pubmed/30017653?tool=bestpractice.com
[122]El Dib R, Gomaa H, Carvalho RP, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2016 Jul 25;(7):CD006663.
http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006663.pub4/full
http://www.ncbi.nlm.nih.gov/pubmed/27454104?tool=bestpractice.com
[123]Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019 Aug;96(2):107-17.
https://onlinelibrary.wiley.com/doi/10.1111/cge.13546
http://www.ncbi.nlm.nih.gov/pubmed/30941742?tool=bestpractice.com
[124]Germain DP, Altarescu G, Barriales-Villa R, et al. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol Genet Metab. 2022 Sep-Oct;137(1-2):49-61.
https://www.sciencedirect.com/science/article/pii/S1096719222003705?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35926321?tool=bestpractice.com
De modo geral, concorda-se que a TRE pode retardar a evolução da lesão orgânica nos rins e no coração, mas esses órgãos podem não retornar à função normal. Os homens devem ser tratados assim que forem diagnosticados; as mulheres devem ser tratadas se apresentarem sintomas de envolvimento de órgãos importantes.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
O início da TRE deve ser considerado em pacientes assintomáticas do sexo feminino com evidências laboratoriais, histológicas ou de imagem de envolvimento renal, cardíaco ou do sistema nervoso central.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
A alfa-agalsidase e a beta-agalsidase beta demonstraram eficácia e segurança em ensaios clínicos randomizados e controlados.[125]Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N Engl J Med. 2001 Jul 5;345(1):9-16.
http://www.nejm.org/doi/full/10.1056/NEJM200107053450102#t=article
http://www.ncbi.nlm.nih.gov/pubmed/11439963?tool=bestpractice.com
[126]Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001 Jun 6;285(21):2743-9.
http://jama.jamanetwork.com/article.aspx?articleid=193887
http://www.ncbi.nlm.nih.gov/pubmed/11386930?tool=bestpractice.com
Os dados dos ensaios e registros demonstraram redução do risco de AVC, melhora nos desfechos cardíacos e renais, melhora da dor e da qualidade de vida e melhora da sintomatologia gastrointestinal.[127]Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008 Feb;94(2):153-8.
http://www.ncbi.nlm.nih.gov/pubmed/17483124?tool=bestpractice.com
[128]Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007 Jan 16;146(2):77-86.
http://www.ncbi.nlm.nih.gov/pubmed/17179052?tool=bestpractice.com
[129]Beck M, Ricci R, Widmer U, et al. Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest. 2004 Dec;34(12):838-44.
http://www.ncbi.nlm.nih.gov/pubmed/15606727?tool=bestpractice.com
[130]Mehta A, Beck M, Elliott P, et al; Fabry Outcome Survey investigators. Enzyme replacement therapy with agalsidase alfa in patients with Fabry's disease: an analysis of registry data. Lancet. 2009 Dec 12;374(9706):1986-96.
http://www.ncbi.nlm.nih.gov/pubmed/19959221?tool=bestpractice.com
[131]Germain DP, Arad M, Burlina A, et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease - a systematic literature review by a European panel of experts. Mol Genet Metab. 2019 Mar;126(3):224-35.
https://www.sciencedirect.com/science/article/pii/S1096719218301926
http://www.ncbi.nlm.nih.gov/pubmed/30413388?tool=bestpractice.com
[132]Sheng S, Wu L, Nalleballe K, et al. Fabry's disease and stroke: effectiveness of enzyme replacement therapy (ERT) in stroke prevention, a review with meta-analysis. J Clin Neurosci. 2019 Jul;65:83-6.
http://www.ncbi.nlm.nih.gov/pubmed/30955952?tool=bestpractice.com
[133]Spada M, Baron R, Elliott PM, et al. The effect of enzyme replacement therapy on clinical outcomes in paediatric patients with Fabry disease - a systematic literature review by a European panel of experts. Mol Genet Metab. 2019 Mar;126(3):212-23.
https://www.sciencedirect.com/science/article/pii/S1096719218301860
http://www.ncbi.nlm.nih.gov/pubmed/29785937?tool=bestpractice.com
Esses agentes são seguros para uso em crianças e têm o mesmo benefício em homens e mulheres. A produção de anticorpos é provocada pela TRE nos homens, mas o impacto disso sobre a eficácia do tratamento é desconhecido. As mulheres não desenvolvem anticorpos, talvez porque sejam heterozigotas e tenham enzimas circulantes. A alfa-agalsidase e a beta-agalsidase são efetivas na prevenção das complicações renais e cardiovasculares, em comparação com a ausência de tratamento. A beta-agalsidase está associada a um menor risco de complicações cerebrovasculares, em comparação com a beta-agalsidase ou a ausência de tratamento.[134]El Dib R, Gomaa H, Ortiz A, et al. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS One. 2017 Mar 15;12(3):e0173358.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0173358
http://www.ncbi.nlm.nih.gov/pubmed/28296917?tool=bestpractice.com
A TRE é eficaz na redução dos escores de dor e das concentrações de globotriaosilceramida no plasma, rim e coração.[122]El Dib R, Gomaa H, Carvalho RP, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2016 Jul 25;(7):CD006663.
http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006663.pub4/full
http://www.ncbi.nlm.nih.gov/pubmed/27454104?tool=bestpractice.com
A pegunigalsidase alfa é outra opção para tratar a doença de Fabry em adultos, e tem a vantagem de uma meia-vida plasmática mais longa.[135]National Institute for Health and Care Excellence. Pegunigalsidase alfa for treating Fabry disease. Oct 2023 [internet publication].
https://www.nice.org.uk/guidance/ta915
A terapia com chaperonas consiste em moléculas pequenas que aumentam a atividade da enzima deficiente em pacientes com atividade enzimática residual.[136]Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471-86.
http://www.ncbi.nlm.nih.gov/pubmed/25587658?tool=bestpractice.com
Esses compostos auxiliam o tráfego intracelular da enzima para melhorar a disponibilidade para o lisossomo a partir do retículo endoplasmático. Ela pode funcionar em combinação com a terapia de reposição enzimática e não é afetada por anticorpos.
O migalastat é uma chaperona oral que aumenta a atividade da enzima endógena alfagalactosidase A em pacientes com uma mutação favorável. Ensaios demonstraram a segurança e a utilidade desta terapia em pacientes com mutações favoráveis, e o tratamento é atualmente licenciado e está disponível nos EUA, Europa, Canadá, Japão e vários outros países.[137]Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016 Aug 11;375(6):545-55.
http://www.nejm.org/doi/full/10.1056/NEJMoa1510198
http://www.ncbi.nlm.nih.gov/pubmed/27509102?tool=bestpractice.com
[138]National Institute for Health and Care Excellence. Migalastat for treating Fabry disease. Feb 2017 [internet publication].
https://www.nice.org.uk/guidance/hst4
[139]Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017 Apr;54(4):288-96. [Erratum in: J Med Genet. 2018.]
https://jmg.bmj.com/content/54/4/288.long
http://www.ncbi.nlm.nih.gov/pubmed/27834756?tool=bestpractice.com
[140]Schiffmann R, Bichet DG, Jovanovic A, et al. Migalastat improves diarrhea in patients with Fabry disease: clinical-biomarker correlations from the phase 3 FACETS trial. Orphanet J Rare Dis. 2018 Apr 27;13(1):68.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0813-7
http://www.ncbi.nlm.nih.gov/pubmed/29703262?tool=bestpractice.com
Os médicos precisam confirmar que a mutação do paciente é favorável. O fabricante aconselha evitá-la se a taxa de filtração glomerular estimada (TFGe) for inferior a 30 mL/minute/1.73 m². O migalastate é adequado tanto para pacientes virgens de tratamento quanto para pacientes que estão mudando de TRE.[141]Bichet DG, Hopkin RJ, Aguiar P, et al. Consensus recommendations for the treatment and management of patients with Fabry disease on migalastat: a modified Delphi study. Front Med (Lausanne). 2023 Sep 1:10:1220637.
https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1220637/full
http://www.ncbi.nlm.nih.gov/pubmed/37727761?tool=bestpractice.com
Distúrbios de MPS
Manejo geral e de suporte
Uma abordagem multidisciplinar é essencial, pois MPS é um distúrbio multissistêmico. A participação de um especialista deve ser obtida precocemente.
Problemas precoces podem surgir com doença cardíaca e respiratória; especial atenção deve ser dada às vias aéreas em todos os momentos.[142]Shinhar SY, Zablocki RN, Madgy DN. Airway management in mucopolysaccharide disorders. Arch Otolaryngol. 2004 Feb;130(2):233-7.
http://www.ncbi.nlm.nih.gov/pubmed/14967758?tool=bestpractice.com
Complicações otorrinolaringológicas incluem infecção otológicas frequentes.[143]Simmons MA, Bruce IA, Penney S, et al. Otorhinological manifestations of mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2005 May;69(5):589-95.
http://www.ncbi.nlm.nih.gov/pubmed/15850680?tool=bestpractice.com
Complicações musculoesqueléticas são comuns e a participação de outros especialistas, como cirurgiões ortopedistas e neurocirurgiões, é essencial.[144]van der Linden MH, Kruyt MC, Sakkers RJ, et al. Orthopaedic management of Hurler's disease after hematopoietic stem cell transplantation: a systematic review. J Inherit Metab Dis. 2011 Jun;34(3):657-69.
http://link.springer.com/article/10.1007/s10545-011-9304-x/fulltext.html
http://www.ncbi.nlm.nih.gov/pubmed/21416194?tool=bestpractice.com
[145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24.
http://www.sciencedirect.com/science/article/pii/S1096719212002740
http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com
Uma complicação com potencial risco de vida, particularmente durante a anestesia, é a compressão da medula espinhal decorrente de estenose na região craniocervical.[145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24.
http://www.sciencedirect.com/science/article/pii/S1096719212002740
http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com
A síndrome do túnel do carpo é frequente, assim como deformidades da giba.[146]Pastores GM, Meere PA. Musculoskeletal complications associated with lysosomal storage disorders: Gaucher disease and Hurler-Scheie syndrome (mucopolysaccharidosis type I). Curr Opin Rheumatol. 2005 Jan;17(1):70-8.
http://www.ncbi.nlm.nih.gov/pubmed/15604908?tool=bestpractice.com
Uma avaliação cardíaca pré-operatória cuidadosa deve ser realizada em todos os pacientes, dado o número de anomalias valvares cardíacas que ocorrem nesse grupo de doenças.
Tratamento específico
O transplante de células-tronco deve ser considerado para os pacientes gravemente afetados e tem valor estabelecido, por exemplo, na MPS I e na MPS VI graves.[147]Boelens JJ. Trends in hematopoietic stem cell transplantation for inborn errors of metabolism. J Inherit Metab Dis. 2006 Apr-Jun;29(2-3):413-20.
http://www.ncbi.nlm.nih.gov/pubmed/16763911?tool=bestpractice.com
A TRE tem benefício estabelecido na MPS I, II, IVA (síndrome de Morquio A), VI e tipo VII (síndrome de Sly).[148]Hendriksz CJ, Burton B, Fleming TR, et al.; STRIVE Investigators. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014 Nov;37(6):979-90.
http://link.springer.com/article/10.1007/s10545-014-9715-6/fulltext.html
http://www.ncbi.nlm.nih.gov/pubmed/24810369?tool=bestpractice.com
[149]Brunelli MJ, Atallah ÁN, da Silva EM. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis type VI. Cochrane Database Syst Rev. 2021 Sep 17;(9):CD009806.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009806.pub3/full
http://www.ncbi.nlm.nih.gov/pubmed/34533215?tool=bestpractice.com
[150]Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2019 Jun 18;6(6):CD009354.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009354.pub5/full
http://www.ncbi.nlm.nih.gov/pubmed/31211405?tool=bestpractice.com
[151]Wikman-Jorgensen PE, López Amorós A, Peris García J, et al. Enzyme replacement therapy for the treatment of Hunter disease: a systematic review with narrative synthesis and meta-analysis. Mol Genet Metab. 2020 Sep - Oct;131(1-2):206-10.
http://www.ncbi.nlm.nih.gov/pubmed/32773276?tool=bestpractice.com
[152]Gomes DF, Gallo LG, Leite BF, et al. Clinical effectiveness of enzyme replacement therapy with galsulfase in mucopolysaccharidosis type VI treatment: systematic review. J Inherit Metab Dis. 2019 Jan;42(1):66-76.
http://www.ncbi.nlm.nih.gov/pubmed/30740728?tool=bestpractice.com
Várias enzimas estão aprovadas para essas indicações. No Reino Unido, o National Institute for Health and Care Excellence recomenda a elosulfase alfa como uma opção para o tratamento da MPS IVA para pessoas de todas as idades, e está disponível apenas sob um acordo comercial.[153]National Institute for Health and Care Excellence. Elosulfase alfa for treating mucopolysaccharidosis type 4A: highly specialised technologies guidance. Apr 2022 [internet publication].
https://www.nice.org.uk/guidance/hst19
Estão em andamento ensaios clínicos de Fase 3 com a vestronidase alfa.[154]NHS; National Institute for Health Research (NIHR). NIHR Innovation Observatory evidence briefing. Vestronidase alfa (UX-003) for mucopolysaccharidosis type VII (MPS 7; Sly syndrome) NIHRIO (HSRIC) ID: 11463. Apr 2017 [internet publication].
http://www.io.nihr.ac.uk/wp-content/uploads/migrated_new/11463-Vestronidase-alfa-UX-003.pdf
[155]ClinicalTrials.gov. A study of UX003 recombinant human beta-glucuronidase (rhGUS) enzyme replacement therapy in subjects with mucopolysaccharidosis type 7, Sly syndrome (MPS 7). Jul 2020 [internet publication].
https://clinicaltrials.gov/ct2/show/NCT02432144
[156]Akyol MU, Alden TD, Amartino H, et al. Recommendations for the management of MPS VI: systematic evidence- and consensus-based guidance. Orphanet J Rare Dis. 2019 May 29;14(1):118.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-019-1080-y
http://www.ncbi.nlm.nih.gov/pubmed/31142378?tool=bestpractice.com
Doença de Pompe
Os cuidados gerais e de suporte para neonatos são multidisciplinares, envolvendo neurologistas, anestesistas e cardiologistas. A TRE é licenciada para crianças e adultos.[157]Stevens D, Milani-Nejad S, Mozaffar T. Pompe disease: a clinical, diagnostic, and therapeutic overview. Curr Treat Options Neurol. 2022 Nov;24(11):573-88.
https://link.springer.com/article/10.1007/s11940-022-00736-1
http://www.ncbi.nlm.nih.gov/pubmed/36969713?tool=bestpractice.com
As revisões sistemáticas não identificaram nenhum ensaio comparando a eficácia e a segurança da terapia de reposição enzimática para a doença de Pompe de início na infância com outra intervenção, nenhuma intervenção ou placebo.[158]Chen M, Zhang L, Quan S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst Rev. 2017 Nov 20;(11):CD011539.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD011539.pub2/full
http://www.ncbi.nlm.nih.gov/pubmed/29155436?tool=bestpractice.com
[159]Joanne M, Skye N, Tracy M. The effectiveness of enzyme replacement therapy for juvenile-onset Pompe disease: a systematic review. J Inherit Metab Dis. 2019 Jan;42(1):57-65.
https://onlinelibrary.wiley.com/doi/10.1002/jimd.12027
http://www.ncbi.nlm.nih.gov/pubmed/30740732?tool=bestpractice.com
Um estudo com 18 participantes comparou duas doses da alglucosidase alfa. O ensaio forneceu evidências de baixa qualidade de que o tratamento em longo prazo com alglucosidase alfa prolongou acentuadamente a sobrevida, bem como a sobrevida sem ventilação, e melhorou a cardiomiopatia (evidências de baixa qualidade); mas a função cardíaca, o desenvolvimento motor e a proporção de crianças sem ventilação invasiva foram semelhantes para ambas as doses com uma duração mediana do tratamento de 2.3 anos. Não houve diferença significativa entre os grupos de doses.[160]Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009 Sep;66(3):329-35.
https://www.nature.com/articles/pr2009210
http://www.ncbi.nlm.nih.gov/pubmed/19542901?tool=bestpractice.com
São necessários ensaios randomizados, devidamente elaborados, para determinar o regime de dose ideal.
A TRE para a doença de Pompe de início tardio está associada a uma melhora significativa na distância percorrida, mas não na força muscular ou na capacidade vital forçada.[161]Sarah B, Giovanna B, Emanuela K, et al. Clinical efficacy of the enzyme replacement therapy in patients with late-onset Pompe disease: a systematic review and a meta-analysis. J Neurol. 2021 Apr 13 [Epub ahead of print].
https://link.springer.com/article/10.1007%2Fs00415-021-10526-5
http://www.ncbi.nlm.nih.gov/pubmed/33851281?tool=bestpractice.com
Mais estudos prospectivos são necessários. A avalglucosidase é uma opção para a doença de Pompe de início tardio em crianças com 1 ano de idade ou mais.[162]Kishnani PS, Diaz-Manera J, Toscano A, et al. Efficacy and safety of avalglucosidase alfa in patients with late-onset Pompe disease after 97 weeks: a phase 3 randomized clinical trial. JAMA Neurol. 2023 Jun 1;80(6):558-67.
https://jamanetwork.com/journals/jamaneurology/fullarticle/2802973#google_vignette
http://www.ncbi.nlm.nih.gov/pubmed/37036722?tool=bestpractice.com
[163]Dalmia S, Sharma R, Ramaswami U, et al. Enzyme replacement therapy for late-onset Pompe disease. Cochrane Database Syst Rev. 2023 Dec 12;12(12):CD012993.
http://www.ncbi.nlm.nih.gov/pubmed/38084761?tool=bestpractice.com
[164]National Institute for Health and Care Excellence. Cipaglucosidase alfa with miglustat for treating late-onset Pompe disease. Aug 2023 [internet publication].
https://www.nice.org.uk/guidance/ta912
A cipaglucosidase alfa é outra opção aprovada para o tratamento da doença de Pompe de início tardio em adultos que não estiverem melhorando com a TRE em uso. A cipaglucosidase alfa só está aprovada para uso em combinação com o miglustate (um estabilizador enzimático).[165]Blair HA. Cipaglucosidase alfa: first approval. Drugs. 2023 Jun;83(8):739-45.
https://link.springer.com/article/10.1007/s40265-023-01886-5
http://www.ncbi.nlm.nih.gov/pubmed/37184753?tool=bestpractice.com
As diretrizes do consenso europeu recomendam um período inicial de 2 anos de TRE para adultos sintomáticos com doença de Pompe. A TRE pode ser mantida durante a gravidez e a lactação. Os músculos esqueléticos e a função respiratória devem ser avaliados durante o tratamento.[166]van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017 Jun;24(6):768-e31.
http://www.ncbi.nlm.nih.gov/pubmed/28477382?tool=bestpractice.com
doença de Tay-Sachs
Somente cuidados paliativos são necessários para a forma infantil.
As formas de início na fase juvenil ou adulta requerem cuidados de suporte, educação quanto a necessidades especiais e avaliação neurológica. Demência e ataxia são complicações em longo prazo que precisam ser consideradas no manejo.
A TRE não está disponível para a doença de Tay-Sachs.
Doenças de Niemann-Pick
Cuidados paliativos são necessários apenas para a forma infantil grave de Niemann-Pick do tipo A. Os cuidados de suporte são indicados para as formas menos graves.
Niemann-Pick do tipo B se apresenta tipicamente em adultos com doença pulmonar e/ou hepatoesplenomegalia. Essa doença é essencialmente intratável. Terapias de suporte devem ser orientadas por avaliações feitas por um pneumologista, gastroenterologista e hematologista.[167]Geberhiwot T, Wasserstein M, Wanninayake S, et al. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann-Pick disease types A, B and A/B). Orphanet J Rare Dis. 2023 Apr 17;18(1):85.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-023-02686-6
http://www.ncbi.nlm.nih.gov/pubmed/37069638?tool=bestpractice.com
Niemann-Pick do tipo C é extremamente variável, mas a avaliação neurológica geralmente é importante. A terapia de redução de substrato (TRS) está disponível para Niemann-Pick do tipo C.[54]Patterson MC, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017 Dec;7(6):499-511.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5800709
http://www.ncbi.nlm.nih.gov/pubmed/29431164?tool=bestpractice.com
A TRS com miglustate demonstrou estabilizar os sintomas neurodegenerativos, incluindo disfagia.[168]Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J Rare Dis. 2018 Aug 15;13(1):140.
https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0844-0
http://www.ncbi.nlm.nih.gov/pubmed/30111334?tool=bestpractice.com
A avaliação neurológica antes da TRS é importante.