História e exame físico
Principais fatores diagnósticos
comuns
presença de fatores de risco
Os principais fatores de risco incluem etnia asquenaze para muitas doenças do armazenamento lisossomal (DALs), e sexo masculino para algumas DALs (mucopolissacaridose II, doenças de Fabry). Alguns estão presentes na infância e outros na fase adulta. A doença de Gaucher do tipo 1 (início na idade adulta), a doença de Niemann-Pick do tipo A e a doença de Tay-Sachs são mais comuns entre os judeus asquenazes.
História familiar
Por serem distúrbios hereditários, devem ser avaliados após a devida consulta a um geneticista clínico. Ocasionalmente os pacientes não têm história familiar porque as famílias são pequenas (estas doenças são principalmente autossômicas recessivas, e os portadores geralmente são assintomáticos) ou porque pode ser uma mutação nova. Uma história familiar na linhagem estendida é comum nas doenças do armazenamento lisossomal ligadas ao cromossomo X (doença de Fabry, mucopolissacaridose [MPS] II e doença de Danon).
início na infância (MPS, doenças de Pompe, Gaucher, Fabry, Niemann-Pick do tipo A)
Idade <1 ano: as formas graves dos distúrbios de mucopolissacaridose (MPS), doença de Pompe clássica e formas neuronopáticas da doença de Gaucher presentes na primeira infância.[30][35][46][50][79] Idade de início precoce é um forte indicador de evolução grave em longo prazo, e frequentemente é associada à baixa atividade enzimática residual.
Idade entre 1 e 10 anos: a maioria das MPS (inclusive variantes atenuadas) e a Niemann-Pick do tipo A se manifestam até essa idade.[80] A maioria dos homens com doença de Fabry clássica terão tido alguns sintomas, embora a idade média ao diagnóstico nos homens seja mais avançada.[38]
início na adolescência (doenças de Fabry, Pompe, Gaucher dos tipos 1 e 3, mucopolissacaridose, Niemann-Pick dos tipos B e C)
Idade entre 10 e 20 anos: a maioria das doenças de Gaucher do tipo 3; a maioria das doenças de Gaucher do tipo 1 mais graves, mas não todas; a maioria das doenças de Fabry mais clássicas e a maioria das doenças de Pompe, inclusive de início na idade adulta, são sintomáticas, embora nem todas sejam diagnosticadas nessas idades. Niemann-Pick dos tipos B e C são variáveis; os fenótipos graves se manifestam mais precocemente, os fenótipos mais leves mais tardiamente. As formas atenuadas dos distúrbios de mucopolissacaridose também são altamente variáveis.
início na idade adulta (doenças de Fabry, Gaucher do tipo 1 e Pompe)
Idade entre 20 e 50 anos: idade típica de início para a maioria dos pacientes com o tipo 1 da doença de Gaucher, e a idade em que os pacientes com doença de Fabry clássica desenvolvem problemas clínicos.
Idade >50 anos: as doenças do armazenamento lisossomal provavelmente não se apresentam em idades avançadas; exceto as formas mais leves da doença de Gaucher do tipo 1, doença de Fabry atípica e alguns pacientes com doença de Pompe mais branda.
hepatomegalia e/ou esplenomegalia
A hepatomegalia é observada na doença de Gaucher dos tipos 2 e 3, nos distúrbios de mucopolissacaridose, nas doenças de Pompe e Niemann-Pick.[5][30][46][49][54]
A esplenomegalia é observada na doença de Gaucher dos tipos 2 e 3, nos distúrbios de mucopolissacaridose e nas doenças de Pompe e Niemann-Pick.[5][30][46][49][54]
A hepatoesplenomegalia é comum nos distúrbios de mucopolissacaridose, nas doenças de Gaucher e Niemann-Pick dos tipos A, B e C.[5][30][46][49][54]
hiperacusia
Típica da doença de Tay-Sachs.[81]
história de insuficiência renal
Encontrada na doença de Fabry na idade adulta.[31]
erupções/lesões cutâneas
Encontradas na doença de Fabry; outras doenças do armazenamento lisossomal.[31][Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) flanco, (B) genitais, (C) umbigo, (D) coluna lombar, (E) pododáctilosOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].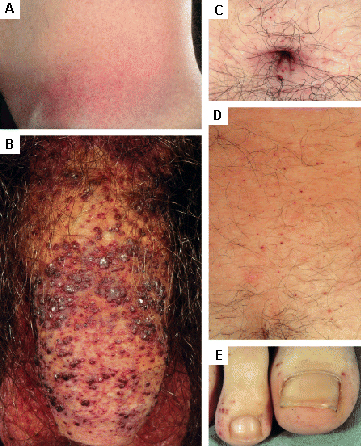 [Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].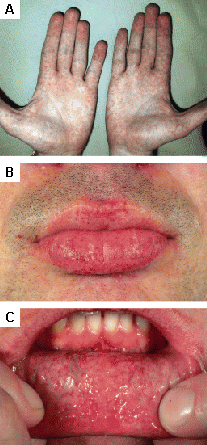
"mancha vermelho-cereja" macular na oftalmoscopia
A coroide vista através da fóvea central aparece como uma área circular vermelha cercada por retina branco-acinzentada. Achado clássico na doença de Tay-Sachs infantil.[56]
atrofia óptica ou retinite pigmentosa na oftalmoscopia
Atrofia óptica ou retinite pigmentosa são observadas na forma juvenil da doença de Tay-Sachs.
opacificação da córnea na oftalmoscopia
Outros fatores diagnósticos
comuns
atraso no neurodesenvolvimento
comprometimento da audição/surdez súbita
Encontrados na doença de Fabry e nos distúrbios de mucopolissacaridose.[49]
catarata na oftalmoscopia
Uma catarata característica é observada na doença de Fabry com vasos retinianos tortuosos. A catarata também é observada na doença de Gaucher, distúrbios da mucopolissacaridose e outras doenças do armazenamento lisossomal.[56]
distúrbios da motilidade ocular
demência progressiva e ataxia ou distúrbio da marcha
Observadas em várias doenças do armazenamento lisossomal de início na fase adulta e juvenil, muitas vezes acompanhadas de ataxia ou distúrbio da marcha (por exemplo, doença de Tay-Sachs juvenil e crônica e doença de Niemann-Pick do tipo C).[54]
retardo do crescimento pôndero-estatural
contratura articular
depressão
Muito comum nos adultos com doença de Fabry.[12] Também encontrada nas doenças de Gaucher, Pompe e Tay-Sachs.
anormalidades esqueléticas, incluindo deformidade espinhal
Sinal inicial frequente nos distúrbios de mucopolissacaridose.[49]
hidrocefalia
Sinal inicial frequente nos distúrbios de mucopolissacaridose.[49]
história de infecções recorrentes do trato respiratório
psicose
distúrbios do movimento
Inespecíficos; porém ataxia, distonia e cataplexia são todas características manifestações de neurodegeneração nas doenças do armazenamento lisossomal (por exemplo, doença de Niemann-Pick do tipo C e Tay-Sachs), e o parkinsonismo pode ocorrer em associação à doença de Gaucher na fase adulta.[30][54]
Incomuns
acidente vascular cerebral prematuro/ataque isquêmico transitório
Observados na doença de Fabry (frequentemente na circulação posterior).[31]
Fatores de risco
Fortes
sexo masculino (mucopolissacaridose [MPS] II, doença de Fabry)
A MPS II é um traço ligado ao cromossomo X; as manifestações são extremamente incomuns no sexo feminino.
A doença de Fabry também é ligada ao cromossomo X, mas as mulheres heterozigotas (>75%) tipicamente têm sintomas, embora menos graves, expressão mais variável e idade de início mais avançada.[36][37][38][39][40]
etnia asquenaze
A taxa de portadores entre judeus asquenazes é cerca de 1 em 15 para a doença de Gaucher, 1 em 30 para Tay-Sachs e 1 em 80 a 1 em 100 para Niemann-Pick do tipo A.[11]
O uso deste conteúdo está sujeito ao nosso aviso legal