Abordagem
As doenças do armazenamento lisossomal (DALs) são um grupo clinicamente diverso de doenças e o diagnóstico diferencial é vasto. As diferentes DALs são facilmente confundidas umas com as outras; o encaminhamento ao especialista em uma etapa precoce é importante.
Já está disponível tratamento para muitas DALs; portanto, há uma ênfase crescente na necessidade de diagnóstico e intervenção precoces antes de ocorrerem danos orgânicos significativos.
O atraso no diagnóstico é uma questão importante para todas as DALs. O diagnóstico de uma DAL deve ser considerado em casos com características clínicas relevantes indicativas de uma DAL.
DALs são distúrbios hereditários, portanto, uma história familiar detalhada deve ser colhida. Deve-se procurar a ajuda de um geneticista clínico se o rastreamento familiar precisar ser realizado.
História e exame físico
O diagnóstico laboratorial e clínico deve ser integrado e a possibilidade de uma DAL deve ser considerada em casos com características clínicas relevantes indicativas de uma DAL.
Doença de Gaucher
A doença de Gaucher do tipo 1 tipicamente se apresenta em adultos com trombocitopenia e/ou esplenomegalia.[30] Fadiga e hepatomegalia são sintomas frequentes.[30] É mais comum em judeus asquenazes.[10][11]
A doença de Gaucher (tipo 2 grave, neuronopática aguda) se apresenta em neonatos com retardo do crescimento pôndero-estatural, dificuldade de alimentação, hepatoesplenomegalia, pele anormal, convulsões e doença grave do sistema nervoso central; esses pacientes geralmente morrem nos primeiros meses de vida.[30]
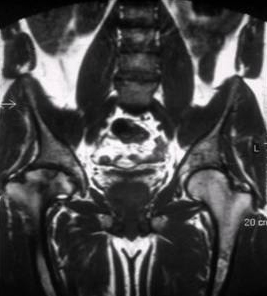
O tipo 3 (neuronopática crônica) se apresenta principalmente em idades entre 10 e 20 anos com distúrbios da motilidade ocular, hepatoesplenomegalia e dor óssea. Convulsões, catarata, valvopatia cardíaca, contraturas articulares e depressão também podem ocorrer.[41][42][Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética do esqueleto na doença de Gaucher do tipo 1 mostrando deposição esquelética disseminada de substrato com necrose associada e infarto ósseo. Há necrose avascular da cabeça femoral (seta)Do acervo pessoal do professor Atul B. Mehta [Citation ends].
O parkinsonismo pode ocorrer em associação com a doença de Gaucher nos adultos.[30]
Pode haver uma história inespecífica de infecções recorrentes do trato respiratório.
Distúrbios da motilidade ocular, como atraso no início dos movimentos sacádicos horizontais, são comuns na doença infantil.[43]
Doença de Fabry
A doença de Fabry muitas vezes passa despercebida na infância.[38]
As características manifestas típicas incluem dor tipo queimação nos membros, febre, dor abdominal e diarreia.[16][17][44] Pode se manifestar na idade adulta com ataque isquêmico transitório ou acidente vascular cerebral (AVC), hipertrofia cardíaca (cardiomiopatia hipertrófica) e insuficiência renal crônica.[31]
Podem ocorrer opacificação da córnea, catarata, hipertensão, hipotireoidismo, valvopatia cardíaca, deficiência auditiva/surdez súbita e depressão.[12]
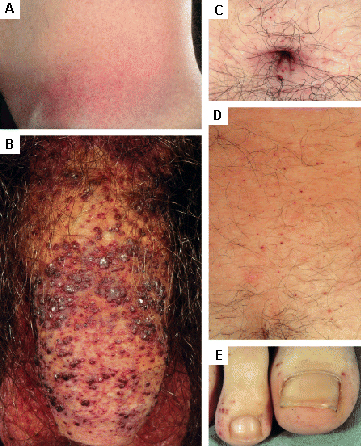
Também são observadas lesões cutâneas.[31] Os exemplos incluem angioqueratoma e telangiectasia.[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) flanco, (B) genitais, (C) umbigo, (D) coluna lombar, (E) pododáctilosOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
 [Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].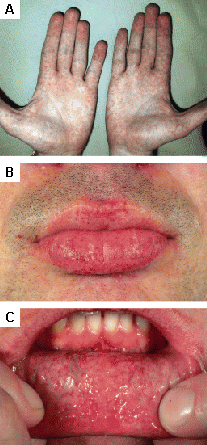
Os sintomas orais incluem agenesia dentária, dentes supranumerários e hipossalivação.[45]
Mulheres heterozigotas frequentemente (>75% dos casos) são sintomáticas.[36][37][38][39][40]
Mucopolissacaridose (formas graves de mucopolissacaridose [MPS] I, II e outras)
MPS tipicamente se apresenta no período neonatal ou na infância/primeira infância com fácies anormal, grande perímetro cefálico, hidrocefalia, aumento da língua, hepatoesplenomegalia, disostose e malformação da coluna vertebral (incluindo deformidade da giba), opacificação da córnea, atraso no neurodesenvolvimento, deformidade articular e retardo do crescimento pôndero-estatural.[5][46]
Também podem ocorrer anomalias cardíacas valvares, dispneia, dificuldade de respiração e deglutição.[47] Comprometimento cognitivo é comum.
Crianças com MPS III podem apresentar sintomas de transtorno do espectro do autismo, incluindo atraso de linguagem e comunicação social comprometida. Os sintomas geralmente surgem na idade avançada do que o transtorno do espectro do autismo idiopático, após um período de desenvolvimento normal.[48]
A MPS também pode se apresentar como hidropisia fetal não imune recorrente.[1]
Pode haver uma história inespecífica de infecções recorrentes do trato respiratório.[49]
Variantes atenuadas se apresentam mais tarde (tipicamente com 5 a 15 anos de idade), mas podem se apresentar em pacientes >30 anos com deformidade articular, síndrome do túnel do carpo, hepatoesplenomegalia, neuropatia compressiva, doença cardíaca e pulmonar, e deficiência auditiva/surdez súbita.
Doença de Pompe
Também conhecida como doença de depósito de glicogênio do tipo II, a forma infantil clássica se apresenta nas primeiras semanas ou meses de vida como dificuldades de alimentação, retardo do crescimento pôndero-estatural, hipotonia e hipertrofia cardíaca com eletrocardiograma (ECG) anormal e alterações ecocardiográficas de cardiomiopatia hipertrófica.[35][50][51]
Podem ocorrer aumento da língua, hepatomegalia e deficiência auditiva.[45]
A morte geralmente ocorre até o primeiro ano de vida sem tratamento específico.
A característica clássica da doença de Pompe é a fatigabilidade manifestada em todas as idades (por exemplo, dificuldades alimentares na primeira infância, desempenho insatisfatório em esportes na infância, dificuldades respiratórias, quedas e dificuldade para subir escadas na fase adulta).
Pode haver uma história inespecífica de infecções recorrentes do trato respiratório.[52]
Variantes atenuadas de início tardio se apresentam dos 10 aos 20 anos de idade (podem chegar a >30 anos) com sintomas de disfunção de músculos esqueléticos: dificuldade de subir escadas, levantar-se/deitar-se, dificuldade respiratória, fadiga, contratura articular e depressão.[53]
Doença de Niemann-Pick
A apresentação depende do tipo: hepatoesplenomegalia nos tipos A, B e C; atraso no neurodesenvolvimento no tipo A; e demência progressiva, ataxia ou distúrbio da marcha ou problemas de audição no tipo C.[54][55]
Os sintomas psiquiátricos precedem os sintomas físicos em 75% dos pacientes com o tipo C. Comprometimentos cognitivos, de memória e instrumentais são os sintomas psiquiátricos mais comuns, seguidos por psicose, comportamento alterado e transtornos do humor.[55]
Distúrbios da motilidade ocular, como paralisias do olhar vertical, são comuns no tipo C.[54]
O tipo A é mais comum em judeus asquenazes.
doença de Tay-Sachs
Na forma infantil, apresenta-se com hiperacusia e uma "mancha vermelho-cereja" macular.[56]
A forma juvenil se apresenta com atrofia óptica (retinite pigmentosa), demência progressiva e ataxia ou distúrbio da marcha, retardo do crescimento pôndero-estatural, contraturas articulares e depressão.
Psicose, ataxia, distonia e cataplexia em adultos jovens podem ser características manifestas de neurodegeneração.[57][58]
Distúrbios da motilidade ocular, como paralisias do olhar vertical, são comuns.
É mais comum em judeus asquenazes.
O diagnóstico confirmatório é feito testando a atividade da enzima específica, ou solicitando sequência do gene e estudos familiares. Em geral, é apropriado para os médicos generalistas considerar o diagnóstico e fazer o rastreamento; no entanto, o encaminhamento precoce ao especialista é recomendado para definir o diagnóstico específico ou descartar DALs se houver qualquer dúvida.
Ensaio enzimático
É a investigação principal para a maioria das DALs. Pode ser apropriado solicitar o ensaio de uma única enzima (por exemplo, glucocerebrosidase se houver suspeita de doença de Gaucher) ou de um grupo de enzimas (por exemplo, solicitar o ensaio enzimático de leucócitos como rastreamento de distúrbios de MPS). Nas DALs ligadas ao cromossomo X (por exemplo, doença de Fabry, MPS II), as mulheres heterozigotas muitas vezes podem ter valores limítrofes, pois cerca de metade de suas células serão normais.
Também é importante solicitar a atividade da enzima plasmática, além da atividade de leucócitos, pois algumas mutações estão associadas à exportação interrompida de enzimas causando redução da atividade plasmática com atividade de leucócitos normais.
Devem-se usar especialistas e laboratórios credenciados, pois os ensaios são tecnicamente complexos e refinamentos são constantemente realizados (por exemplo, para melhorar o diagnóstico de doença de Pompe).[59] Diretrizes para o diagnóstico laboratorial de MPS VI estão disponíveis.[60] Essas diretrizes recomendam cautela no uso da análise isolada de glicosaminoglicanos (GAGs) urinários na confirmação do diagnóstico de MPS VI, e reconhecem a análise da atividade da enzima como um componente fundamental do diagnóstico.
Níveis de substrato
Níveis elevados de substrato apropriado serão detectáveis na presença de uma deficiência enzimática. Portanto, GAGs urinários são elevados nos distúrbios de MPS e oligossacarídeos urinários são elevados na gangliosidose GM1 e GM2.[61] Os níveis urinários de globotriaosilceramida (Gb3) estão elevados na doença de Fabry. Os níveis de tetrassacarídeo de glicose (Glc4) urinário e tetrassacarídeo de glicose (Hex4) plasmático estão aumentados nos pacientes com doença de Pompe.[62] Os níveis plasmáticos de glicosilceramida estão elevados na doença de Gaucher.
análise do ácido desoxirribonucleico (DNA)
Estudos genéticos podem fornecer confirmação do diagnóstico para a maioria das DALs.
Na doença de Gaucher, há 6 mutações comuns observadas entre judeus asquenazes, e um pequeno número de outras mutações recorrentes ocorre de tal forma que há kits diagnósticos disponíveis que permitem a detecção de mutações comuns por reação em cadeia da polimerase.[63][64] Muitas vezes, apenas um alelo mutante é detectado; portanto, as manifestações clínicas devem ser consideradas para diferenciar portadores daqueles que sofrem da doença.
Na doença de Fabry, a maioria dos homens classicamente afetados apresentam mutações "privadas" (mutações raras geralmente encontradas apenas em populações pequenas), as quais tipicamente são nulas (associadas à ausência da enzima).[31]
Na doença de Pompe, nos distúrbios de MPS e na maioria das outras DALs, um grande número de mutações é relatado para cada doença, mas há mutações recorrentes que tipicamente são mais comuns em algumas comunidades que em outras.
O resultado do DNA precisa ser considerado com base em informações clínicas detalhadas e dados sobre atividade enzimática.
Algumas mutações são polimorfismos e podem não estar associadas à doença. Muitos testes de reação em cadeia da polimerase rastreiam apenas mutações comuns; mutações "privadas" são detectadas somente pela análise da sequência em laboratórios de pesquisa.
Cromatografia líquida desnaturante de alta pressão é um método para rastreamento rápido de mutações de base única.
A detecção do portador em distúrbios ligados ao cromossomo X pode ser feita de modo confiável somente com análise do DNA.
Nem todas as mutações foram analisadas, e alguns distúrbios serão diagnosticados somente em laboratórios especializados; é fundamental consultar centros especializados ao desenvolver e implementar a estratégia diagnóstica dessas doenças raras.
Hemograma completo
A anemia pode ser multifatorial decorrente de doença crônica, comprometimento renal, dificuldades alimentares ou substituição da medula óssea.
A leucopenia tipicamente é decorrente de esplenomegalia. Inclusões anormais podem ser observadas nos leucócitos: por exemplo, linfócitos positivos para ácido periódico de Schiff na doença de Pompe; aparência anormal dos leucócitos no sangue/medula óssea na doença de Tay-Sachs.[65]
Trombocitopenia tipicamente é decorrente de esplenomegalia; no entanto, pode haver função plaquetária comprometida, por exemplo, na síndrome de Hermansky-Pudlak.[66]
ECG e ecocardiografia
São essenciais para avaliação cardíaca na doença de Pompe infantil, na doença de Fabry em adultos e em muitos distúrbios neonatais de MPS.[67][68] Achados podem indicar câmaras cardíacas aumentadas, valvas anormais e defeitos funcionais nesses pacientes.
Testes de função pulmonar
São fundamentais nos tipos A e B da doença de Niemann-Pick, para avaliar a gravidade. Função pulmonar anormal com transferência gasosa deficiente serão detectadas nesses pacientes.[69]
Exame oftalmológico
"Mancha cereja" macular e atrofia óptica ou retinite pigmentosa são observadas na doença de Tay-Sachs.[56] Uma opacificação da córnea é observada nos distúrbios de MPS, na doença de Fabry e em algumas formas da doença de Gaucher.[43] Catarata característica é observada na doença de Fabry com vasos retinianos tortuosos.[56] Cataratas também são observadas na doença de Gaucher, distúrbios de MPS e outras DALs.
Biópsia do tecido afetado
Deve ser considerada somente após a realização de testes menos invasivos. A biópsia de tecido de um órgão aumentado e/ou com função alterada (por exemplo, fígado, rim, coração) pode ser realizada para demonstrar níveis elevados de substrato, muitas vezes associados ao ingurgitamento celular.
A biópsia de medula óssea frequentemente é realizada na investigação de uma hepatoesplenomegalia, e pode revelar células de depósito características, como macrófagos carregados de substrato na doença de Gaucher.[70] A microscopia eletrônica do tecido afetado pode mostrar alterações características de danos lisossomais. A biópsia muscular é de utilidade diagnóstica em muitas DALs (por exemplo, doença de Pompe de início tardio; no entanto, as aparências podem ser normais); a biópsia de pele muitas vezes é preferencial por ser relativamente não invasiva e os fibroblastos da pele cultivados são úteis para o ensaio enzimático.[71][72][73] A decisão sobre o tecido adequado para a biópsia será tomada pelo especialista.[Figure caption and citation for the preceding image starts]: Aspirado da medula óssea mostrando célula de Gaucher típica. Este é um macrófago que ingeriu material celular; o substrato não degradado (glicosilceramida) se acumula nos lisossomosDo acervo pessoal do professor Atul B. Mehta [Citation ends].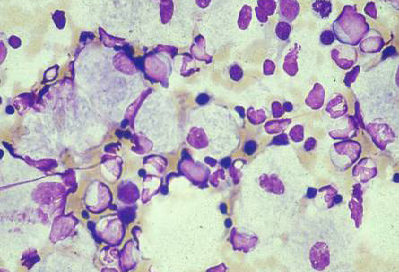 [Figure caption and citation for the preceding image starts]: Imagem de microscópio eletrônico de biópsia de células epiteliais pulmonares na doença de Fabry mostrando depósitos característicos de substrato em lisossomos, formando "corpos em zebra": (A) aumento de 8000x, (B) aumento de 62,500xKelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. 2000 Aug;55(8):720-1; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Imagem de microscópio eletrônico de biópsia de células epiteliais pulmonares na doença de Fabry mostrando depósitos característicos de substrato em lisossomos, formando "corpos em zebra": (A) aumento de 8000x, (B) aumento de 62,500xKelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. 2000 Aug;55(8):720-1; usado com permissão [Citation ends].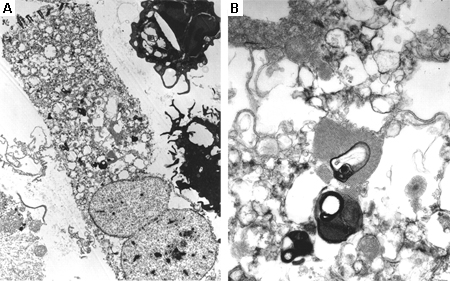
Exames por imagem
As DALs são distúrbios multissistêmicos diversos, e muitas formas diferentes de exames de imagem são importantes e relevantes. São exemplos a tomografia computadorizada (TC) e a ressonância nuclear magnética (RNM) para medir os volumes dos órgãos e avaliar o esqueleto na doença de Gaucher; ecocardiografia, ultrassonografia ou RNM para avaliar coração, rins e cérebro na doença de Fabry; e TC e radiografia para avaliar hidrocefalia comunicante, displasia atlanto-axial e anomalias da coluna espinhal/vertebral nos distúrbios de MPS.[42][74][75][76][77][Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética do esqueleto na doença de Gaucher do tipo 1 mostrando deposição esquelética disseminada de substrato com necrose associada e infarto ósseo. Há necrose avascular da cabeça femoral (seta)Do acervo pessoal do professor Atul B. Mehta [Citation ends].
Significado de resultados negativos do exame
O encaminhamento precoce a um especialista é necessário. O médico deve manter uma estreita ligação com o laboratório e garantir que as amostras cheguem ao laboratório rapidamente para evitar a deterioração e a possível interpretação de falsos-negativos. Alguns ensaios estão sendo refinados (por exemplo, para a doença de Pompe); alguns testes de DNA/enzimas podem estar disponíveis apenas por encaminhamento, pelo especialista, a laboratórios de pesquisa internacionais.[78] A deficiência de uma proteína ativadora de esfingolipídeos ou saposina pode resultar em um fenótipo clínico que se assemelha a uma DAL e pode estar por trás da doença de Gaucher e da leucodistrofia metacromática.[1]
O uso deste conteúdo está sujeito ao nosso aviso legal