Etiologia
A síndrome de Alport é uma entre várias doenças causadas por mutações nos genes que codificam o colágeno tipo IV. Ela se deve a mutações nos genes COL4A3, COL4A4 e COL4A5.[1] Eles codificam as cadeias alfa-3, alfa-4 e alfa-5 do colágeno tipo IV, que é um componente importante da membrana basal glomerular. A membrana basal glomerular é única devido à sua espessura (300 a 350 nanômetros) e à sua posição entre duas camadas de células, podócitos e células endoteliais. Outros componentes da membrana basal glomerular são laminina, nidogênio e proteoglicanos de sulfato de heparina. Cada molécula de colágeno tipo IV é composta por três subunidades ou cadeias alfa que se entrelaçam para formar uma estrutura helicoidal. Essas cadeias de colágeno são caracterizadas por uma sequência Gly-X-Y repetida com um domínio não colagenoso no terminal C. Seis genes diferentes, COL4A1 a COL4A6, codificam as diferentes isoformas da cadeia alfa, alfa-1(IV) a alfa-6(IV). Alfa-1(IV) e alfa-2(IV) são expressas livremente, enquanto alfa-3(IV), alfa-4(IV) e alfa-5(IV) são restritas à membrana basal glomerular, membrana basal tubular distal, membrana de Descemet (membrana hialina fina entre a substância própria e a camada endotelial da córnea) e a membrana de Bruch (camada interna da coroide, separando-a da camada pigmentar da retina), cápsula do cristalino anterior, pulmão e cóclea. Isso explica as características fenotípicas observadas na síndrome de Alport nos rins, nos ouvidos e nos olhos. Alfa-6(IV) é encontrada na membrana basal epidérmica.
Os genes COL4 são encontrados como pares em uma orientação "cabeça com cabeça", COL4A1 a COL4A2 no cromossomo 13, COL4A3 a COL4A4 no cromossomo 2 e COL4A5 a COL4A6 no cromossomo X. Mutações em COL4A1 e COL4A2 estão associadas à doença dos pequenos vasos no cérebro que se manifesta como porencefalia (OMIM 120130), enquanto mutações em COL4A1 também causam outras doenças, incluindo angiopatia hereditária com nefropatia, aneurismas e cãibras musculares.[19] Somente as mutações em COL4A3, COL4A4 e COL4A5 têm sido descritas na síndrome de Alport, bem como uma deleção de gene contígua de COL4A5 a COL4A6.[15] As mutações causam perda de expressão ou montagem indevida das cadeias alfa e, assim, falha de montagem e amadurecimento de uma rede normal de colágeno da membrana basal.
Outros fenótipos associados a variantes em COL4A3, COL4A4 e COL4A5 incluem hematúria dismórfica persistente, proteinúria persistente, síndrome nefrótica resistente a esteroides, glomeruloesclerose segmentar focal e doença renal em estágio terminal de causa desconhecida.[16]
[Figure caption and citation for the preceding image starts]: Classificações da síndrome de Alport discutidas neste tópico com mutações genéticas associadasCriado pelo BMJ Knowledge Center com informações fornecidas por Richard N. Sandford [Citation ends].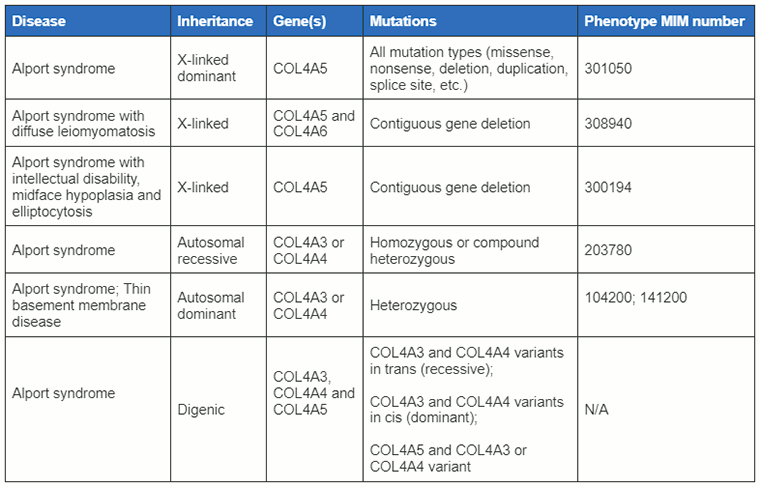
Fisiopatologia
Durante o desenvolvimento precoce, as isoformas alfa-1(IV) e alfa-2(IV) predominam na membrana basal glomerular antes da mudança realizada principalmente para alfa-3(IV), alfa-4(IV) e alfa-5(IV). Acredita-se que essas isoformas melhoram a capacidade da membrana basal glomerular de resistir à degradação proteolítica. A perda dessas cadeias também causa um aumento compensatório na expressão dos outros colágenos tipo IV, tipo V e tipo VI. Portanto, as anormalidades nesses componentes causam danos ultraestruturais à membrana basal, que se manifestam como afinamento, espessamento, divisão e lamelação, as características clássicas observadas em microscopia eletrônica. Supostamente, isso resulta na perda da permeseletividade normal da membrana basal glomerular e no desenvolvimento subsequente de hematúria, proteinúria, glomeruloesclerose e nefrite intersticial.
Classificação
Classificação genética
A síndrome de Alport é uma doença geneticamente heterogênea causada por mutações em genes que codificam diferentes cadeias alfa do colágeno tipo IV, nomeadamente COL4A3, COL4A4 e COL4A5.[1][5] É um dos transtornos hereditários caracterizados por anormalidades na membrana basal glomerular. Aproximadamente 85% dos casos são hereditários, como um caráter ligado ao cromossomo X, e ocorrem devido a mutações no gene COL4A5 localizado em Xq22.3. Os 15% restantes dos casos são autossômicos recessivos e ocorrem devido a mutações nos genes COL4A3 e COL4A4. Os casos autossômicos dominantes raros e autossômico digênicos também ocorrem devido às mutações nos genes COL4A3 e COL4A4.
Síndrome de Alport ligada ao cromossomo X (SAX)
A SAX é o tipo mais comum da síndrome de Alport.[4] Todos os pacientes do sexo masculino têm hematúria e desenvolvem doença renal crônica. Há uma heterogeneidade alélica considerável. Correlações entre genótipo e fenótipo foram descritas com 90% dos pacientes masculinos que sofrem de mutações de perda de função (grandes deleções, mutações nonsense e de deslocamento de quadro) e que atingem a doença renal crônica até os 30 anos de idade.[4][6]
A hematúria se desenvolve em mais de 95% das mulheres com SAX, com algumas desenvolvendo doença renal crônica mais tarde na vida.[7] A história familiar pode estar ausente em cerca de 10% dos casos, sugerindo uma mutação de novo.
Síndrome de Alport autossômica recessiva (SAAR)
A SAAR em uma família é sugerida pela presença da doença precoce grave em homens e mulheres, hematúria isolada em ambos os pais e consanguinidade parental.[8]
Síndrome de Alport autossômica dominante (SAAD)
A SAAD é rara e sugerida por um fenótipo relativamente leve e progressão lenta para doença renal crônica nos pacientes afetados.[9] O termo diagnóstico SAAD não é universalmente aceito para indivíduos heterozigotos para uma única variante COL4A3 ou COL4A4, pois a maioria terá apenas hematúria e um risco muito baixo de desenvolver doença renal crônica e, como resultado, pode não atender aos critérios para a síndrome de Alport. Os filhos de indivíduos com SAAD heterozigotos também não apresentam 50% de probabilidade de desenvolver a síndrome de Alport, o que está implícito no termo “autossômico dominante”.[10]
Síndrome de Alport digênica autossômica
Isso é raro e possui um fenótipo entre ARA e SAAD[11]
Síndrome de Alport com leiomiomatose difusa (SALD)
Na SALD, leiomiomas genitais são frequentemente observados em mulheres. Pode haver também uma história infantil de catarata, disfagia, tosse, alteração no hábito intestinal ou bronquite recorrente. No entanto, frequentemente não há história familiar.
A deleção em COL4A5 e COL4A6 é detectada por amplificação de múltiplas sondas dependentes de ligação e/ou hibridização in situ fluorescente (FISH) (se a deleção for grande).
Síndrome de Alport com dificuldades de aprendizagem
Classificada como a presença de nefropatia com dificuldades de aprendizagem. Normalmente não há história familiar, mas a aparência facial característica de hipoplasia de face média é observada junto com eliptocitose no esfregaço.[12]
A microdeleção do gene COL4A5 inteiro é detectada por FISH.
O uso deste conteúdo está sujeito ao nosso aviso legal