Abordagem
Uma anamnese detalhada é extremamente importante, pois as crises epilépticas geralmente não são presenciadas por médicos. Uma descrição do início da crise é fundamental (para ajudar a categorizar como crise de início focal ou generalizado) e informações adicionais como: o ambiente, qualquer possível fator precipitante, sinais de alerta que a criança ou o jovem talvez seja capaz de narrar detalhadamente e qualquer fenômeno pós-ictal. Um eletroencefalograma (EEG) é usado para ajudar a determinar a síndrome epiléptica específica e identificar características como a fotossensibilidade.[18][19] Exames laboratoriais padrão, como hemograma completo e perfil metabólico básico (glicemia, cálcio, magnésio) não são indicados rotineiramente, mas podem ser solicitados inicialmente para descartar quaisquer causas subjacentes. Exames de neuroimagem podem ser indicados se o início da convulsão não tiver sido testemunhado ou se houver sugestão de um início focal; porém, na maioria dos casos de epilepsias generalizadas com um claro diagnóstico de síndrome eletroclínica, esses exames não são indicados. A testagem genética é recomendada se houver suspeita de certas síndromes epilépticas.
A Comissão de Classificação e Terminologia da ILAE desenvolveu um manual de diagnóstico online das epilepsias para ajudar os médicos a diagnosticarem o(s) tipo(s) de convulsão, diagnosticar síndromes epilépticas e definir a etiologia da epilepsia. ILAE: EpilepsyDiagnosis.org Opens in new window
História
A história pode ser extraída dos pais ou de qualquer testemunha ocular. A história familiar de convulsões ou epilepsia, a história médica pregressa, a história do nascimento e a história do desenvolvimento da criança devem ser observadas.
É importante percorrer os eventos em ordem cronológica, explorando as circunstâncias da convulsão, o que a criança estava fazendo no momento, possíveis fatores desencadeantes (por exemplo, luz, ruídos, privação do sono, fadiga, estado emocional), presença de aura ou outros avisos (por exemplo, déjà vu, tontura, visão alterada) e como as convulsões começaram e como ocorreram. A gravação em vídeo de um episódio ou a captura do evento em um smartphone é muito útil para o médico. Como alternativa, demonstrar alguns dos movimentos ou pedir a qualquer testemunha para imitar o que ela viu costuma ser extremamente útil na determinação do tipo de convulsão (ou seja, mioclônica, atônica, tônica e/ou clônica ou ausência). Tradicionalmente, pensa-se que o seguinte significa uma crise epiléptica: movimentos específicos; tônus; estado de consciência; presença de incontinência, mordida de língua ou movimentos oculares, bem como a forma como a convulsão terminou e se houve quaisquer sinais/sintomas (por exemplo, sonolência, cefaleia, amnésia, confusão) no estado pós-ictal. No entanto, estes por si só não equivalem a uma pessoa que teve uma crise epiléptica.
Tipos de convulsão
As características dos tipos de convulsão incluem:[2]
Ausência: comprometimento (geralmente breve) da consciência, que pode estar associado à imobilidade ou a movimentos estereotipados ou automatismos. É importante diferenciar os dois tipos de crises de ausência, pois ocorrem em diferentes síndromes epilépticas:
Crise de ausência típica: apatia ou olhar perdido, com duração de 5 a 10 segundos, interrompendo as atividades antes normais; pode ser induzida por hiperventilação
Crises de ausência atípicas: início e término menos diferenciados; geralmente não precipitadas por hiperventilação.
Mioclônica: espasmos musculares breves e arrítmicos.
Clônica: espasmos musculares rítmicos com ou sem comprometimento da consciência.
Tônica: extensão ou flexão tônica dos membros.
Tônico-clônica: a fase tônica envolve a perda de consciência do paciente, que possivelmente cai no chão, e a extensão ou flexão dos membros, e pode ser precedida por aura; a fase clônica consiste geralmente em contrações musculares violentas e tremor.
Atônica: breve perda de tônus muscular, causando as conhecidas "síncopes", quando o paciente cai no chão.
A duração das convulsões pode variar de um evento para outro na mesma pessoa, mas a duração também pode ser uma característica específica de determinado tipo de convulsão. Por exemplo, as crises de ausência duram comumente até 20 segundos. Uma duração maior é tão excepcional que justificaria o questionamento do diagnóstico.[20] A maioria das crises tônico-clônicas generalizadas dura <5 minutos. Diferenciar essas crises das crises tônico-clônicas generalizadas que apresentam uma maior duração não é útil em termos de diagnóstico, mas ajuda a tomar decisões imediatas de manejo.
Às vezes, as convulsões podem ser induzidas pela luz. Esse fenômeno é chamado de fotossensibilidade e é uma característica diagnóstica útil. As seguintes síndromes são conhecidas por serem fotossensíveis: epilepsia com mioclonia palpebral, epilepsia mioclônica juvenil, epilepsia com crises tônico-clônicas generalizadas isoladas, síndrome de Dravet e epilepsias mioclônicas progressivas.[21][22]
Exame físico
Em uma convulsão sem fatores precipitantes, a possibilidade de um distúrbio subjacente deve ser considerada durante a avaliação clínica. O médico deve procurar sinais neurocutâneos, pois os quadros clínicos com esses sinais podem se manifestar com convulsões, incluindo o complexo da esclerose tuberosa (ou seja, máculas despigmentadas, marcas de Shagreen, fibromas periungueais, adenoma sebáceo e máculas hipomelanóticas [em formato de folha]), a neurofibromatose do tipo 1 (ou seja, manchas café-com-leite, sardas axilares e inguinais e neurofibromas plexiformes) e a síndrome de Sturge-Weber (por exemplo, hemagiomas [manchas vinho do porto] no rosto ou no tronco).[12]
Um exame neurológico completo é uma parte importante da avaliação. Pressão arterial, medição do perímetro cefálico e um ECG são necessários para qualquer criança que já apresentou convulsão generalizada. Na maioria dos pacientes com convulsões generalizadas recorrentes não há anormalidades.
Eletroencefalograma (EEG)
O EEG é uma investigação padrão para o diagnóstico de epilepsia.[23] As descargas epilépticas podem ser observadas no registro interictal, e os ritmos anormais podem ser característicos de uma síndrome epiléptica específica.[18][19] Cerca de 5% das crianças sem epilepsia apresentam anormalidades inespecíficas no EEG e até 40% das crianças com epilepsia apresentam EEG interictais normais. Portanto, o EEG complementa técnicas de avaliação da história, do estado físico e outras. Um EEG interictal normal não descarta o diagnóstico de epilepsia. A atividade epileptiforme em um registro combinado de despertar e sono aumenta a probabilidade de o diagnóstico de epilepsia estar correto de 0.17 (para crianças sem essas anormalidades) a 0.95.[24]
A hiperventilação, a estimulação fótica e a privação do sono podem ser usadas para provocar convulsões como parte da investigação do EEG ou para realçar as anormalidades no EEG. Na epilepsia do tipo ausência da infância, induzir uma convulsão no consultório ao pedir para a criança hiperventilar por até 3 minutos é uma ferramenta diagnóstica útil, pois resultará em uma crise de ausência em 90% das crianças com epilepsia tipo ausência.[20]
Um vídeo-EEG pode ser útil. Esse recurso é usado para determinar se as convulsões clinicamente observadas são epileptiformes por natureza. Ele também pode ajudar a caracterizar melhor as convulsões e esclarecer o diagnóstico. Alguns estudos sugerem que um EEG é útil para predizer o risco de recorrência de convulsões em crianças e adolescentes.[25][26]
Eletrocardiograma (ECG)
Um ECG deve ser realizado em todas as crianças que tenham uma primeira convulsão para descartar as causas cardíacas de eventos clínicos, principalmente a síndrome do QT longo.[27][28] Outras causas possíveis incluem a síndrome de Brugada e possíveis arritmias. Um cardiologista deve ser consultado se houver dúvida ou se houver história familiar de morte precoce inexplicável.
Neuroimagem
A combinação de achados típicos de EEG que se correlacionam com o quadro clínico geralmente pode ser diagnóstica. No entanto, quando ainda há dúvidas sobre o diagnóstico ou suspeita de causas secundárias, a ressonância nuclear magnética (RNM) é a técnica de imagem definitiva em crianças, pois não envolve a radiação ionizante, e as imagens proporcionam mais detalhes que as tomografias computadorizadas (TCs).
A TC desempenha um papel na investigação aguda de crianças com uma história recente de convulsões e nas quais é preciso descartar a presença de hemorragia intracerebral. Esse exame também é usado quando há uma suspeita de lesão com efeito de massa e a RNM não está disponível.[23][29]
A RNM funcional pode ser usada para mapeamentos pré-operatórios.[30]
Investigações laboratoriais
Estes não são indicados rotineiramente para pacientes com convulsões recorrentes. No entanto, o nível glicêmico, o perfil metabólico básico e um hemograma completo podem ser considerados em crianças com suspeita de fatores precipitantes, como hipoglicemia, anormalidades eletrolíticas, distúrbios metabólicos ou infecção. A medição da glicose sanguínea é uma parte obrigatória da avaliação de toda criança com um nível de consciência reduzido. Recomenda-se que estes testes sejam solicitados pelo menos uma vez para todos os pacientes com convulsões recorrentes.
Teste genético
O teste genético é recomendado para crianças com suspeita de síndrome de Dravet. Também deve ser considerado para suspeita de encefalopatias epilépticas e de desenvolvimento (incluindo suspeita de encefalopatia epiléptica e de desenvolvimento infantil precoce, síndrome de espasmo epiléptico infantil e síndrome de Lennox-Gastaut) ou se anormalidades estruturais sugerirem uma causa genética.[21][31][32][33]
Diagnóstico de síndromes epilépticas específicas
A história do paciente (incluindo tipos de convulsão), EEG, exame físico e informações diagnósticas complementares podem ajudar a classificar a epilepsia em síndromes epilépticas eletroclínicas específicas. Algumas síndromes terão um EEG normal, especialmente na apresentação inicial, portanto a história e a apresentação também são vitais. A identificação de síndromes epilépticas fornece orientação sobre manejo, desfechos, comorbidades e possíveis avaliações adicionais. A idade do paciente também pode orientar quanto ao diagnóstico, pois a incidência de síndromes específicas atinge a intensidade máxima em determinadas faixas etárias.[1]
Início na primeira infância (idade 1 mês a 2 anos)[31]
Encefalopatia epiléptica e de desenvolvimento infantil precoce (EIDEE; inclui síndrome de Ohtahara)
O início geralmente ocorre aos 3 meses de idade (ajustado para prematuridade).
As crises tônicas e/ou mioclônicas são típicas, mas outros tipos de convulsões podem ocorrer.
O EEG pode mostrar padrão surto-supressão, espícula/espícula-onda/ondas agudas multifocais com ou sem lentidão, descontinuidade e/ou lentificação difusa. O padrão surto-supressão compreende surtos de alta voltagem de espículas mistas e ondas agudas e lentas com duração de 1 a 5 segundos, alternando com períodos de supressão acentuada com duração de 3 a 10 segundos, e pode ser observado quando o paciente está acordado ou dormindo. A dessincronização é observada durante espasmos tônicos.
Comportamento ou desenvolvimento neurológico anormal pode estar presente antes do início das convulsões (mas isso pode ser difícil de avaliar).
Variantes genéticas patogênicas causais podem ser identificadas em mais da metade dos pacientes.
Síndrome de espasmos epilépticos infantis (IESS):
O início dos espasmos epilépticos geralmente ocorre entre 1 e 24 meses de idade (pico entre 3 e 12 meses), mas pode ocorrer mais tarde.
Caracterizada por espasmos epilépticos flexores, extensores ou mistos, que geralmente ocorrem de forma agrupada.
EEG: a hipsarritmia (espículas assimétricas assíncronas e ondas lentas) é geralmente, mas nem sempre, observada em registros interictais.
O desenvolvimento pode ou não ser normal antes do início, mas a desaceleração, a parada ou a regressão do desenvolvimento são observadas após o início.
Um subgrupo com a tríade de espasmos infantis, estagnação ou regressão do desenvolvimento e hipsarritmia no EEG é definido como síndrome de West.
Muitas variantes genéticas patogênicas foram associadas à IESS.
Epilepsia mioclônica do lactente (EML):
O início geralmente ocorre entre os 4 meses e os 3 anos (pico entre 6 e 18 meses).
Caracterizada por convulsões mioclônicas que ocorrem várias vezes ao dia, durante o sono e quando acordado. Elas podem ser desencadeadas por ruído, susto, toque ou (menos comumente) estimulação fótica. As convulsões podem ser breves e singulares ou ocorrer de forma agrupada.
EEG: a base é normal, mas descargas de complexos espícula ou poliespícula onda lenta generalizados são observadas durante crises mioclônicas.
O desenvolvimento geralmente é normal antes do início das convulsões. A maioria dos pacientes apresenta desfecho de desenvolvimento normal, mas podem ser observados deficit intelectual leve ou problemas comportamentais. O deficit intelectual moderado a grave é raro.
Síndrome de Dravet:
Início por volta de 1 ano de idade, com crises características prolongadas, febris e afebris, clônicas focais (geralmente hemiclônicas) ou clônicas generalizadas.
Outros tipos de convulsão (por exemplo, crises mioclônicas, atônicas, tônico-clônicas e/ou crises de ausência atípicas) podem aparecer entre as idades de 1 e 4 anos.
O EEG é inicialmente normal. Após os 2 anos de idade, a lentidão é típica e as descargas interictais são frequentemente focais, multifocais e generalizadas. Os registros ictais dependem do tipo de crise.
As crianças normalmente apresentam desenvolvimento normal antes do início das convulsões. Platô de desenvolvimento e outras anormalidades neurológicas, como ataxia, frequentemente se apresentam durante o segundo ano de vida.
Frequentemente, há uma história familiar de convulsões febris. Uma variante patogênica no gene SCN1A está presente em mais de 80% dos casos.
Início na infância (2-12 anos)[7][32]
Epilepsia com crises mioclônico-atônicas (EMAtS; anteriormente conhecida como síndrome de Doose):
Deve ser considerada em crianças em idade pré-escolar que apresentam convulsões generalizadas (tônico-clônicas generalizadas, mioclônicas, ausência) que desenvolvem convulsões mioclônico-atônicas.
Durante uma convulsão mioclônico-atônica, a criança apresenta um breve espasmo (mioclonia), às vezes acompanhado por um grunhido audível, e depois perde abruptamente o controle muscular e pode cair no chão. Há uma perda da consciência associada, mas muito breve (<3 segundos).
O EEG é inicialmente normal, embora mais comumente o registro interictal mostre alguma lentidão com excesso de atividade teta. A aparência do EEG ictal depende do tipo de convulsão: as ausências estão associadas a complexos espícula-onda lenta, as crises tônico-clônicas estão associadas a poliespículas generalizadas de 10 a 15 Hz, e as crises mioclônicas e atônicas estão associadas a descargas de complexos espícula-onda generalizados e irregulares.[Figure caption and citation for the preceding image starts]: Eletroencefalograma (EEG): epilepsia com crises mioclônico-atônicasCortesia do Professor Eric Kossoff; usado com permissão [Citation ends].
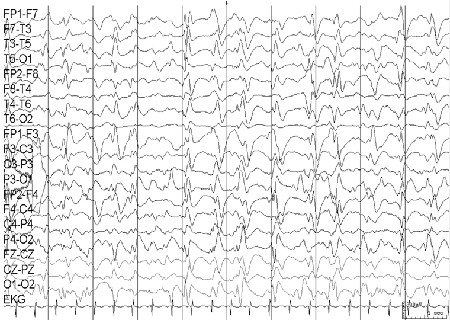
A maioria das crianças afetadas apresenta desenvolvimento normal antes do início da epilepsia, mas cerca de 50% desenvolvem dificuldades de aprendizagem ou deficiência intelectual leve/moderada.[34]
Síndrome de Lennox-Gastaut (SLG):
O início geralmente ocorre entre os 18 meses e os 8 anos, com pico de início aos 3-5 anos.
A presença de convulsões tônicas com duração de 3 segundos a 2 minutos é obrigatória para o diagnóstico.
Muitos outros tipos de convulsão podem ocorrer, incluindo ausência tônico-clônica, atônica, mioclônica, focal e atípica.
Muitos pacientes apresentam apenas alguns dos aspectos característicos durante um determinado período de tempo; as manifestações clínicas muitas vezes não estão todas presentes em uma determinada pessoa.
EEG: o EEG interictal anormal é uma marca registrada desta síndrome, mostrando complexos de espícula-onda lenta difusos (<2.5 Hz) e atividade paroxística rápida generalizada.[Figure caption and citation for the preceding image starts]: Eletroencefalograma (EEG): síndrome de Lennox-GastautCortesia do Professor Eric Kossoff; usado com permissão [Citation ends].
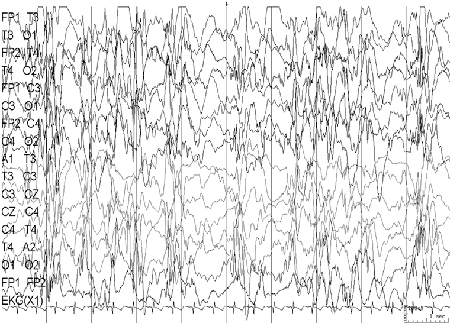
A maioria das crianças apresenta um atraso no desenvolvimento.
Até 75% dos pacientes apresentam episódios recorrentes de estado de mal epiléptico não convulsivo, cujo reconhecimento e tratamento podem ser difíceis.
Variantes patogênicas em muitos genes, bem como uma série de anormalidades cromossômicas e variantes no número de cópias, têm sido associadas à SLG.
Epilepsia com ausência mioclônica (EMA):
O início geralmente ocorre por volta dos 7 anos de idade (intervalo de 1 a 12 anos).
As crises de ausência mioclônica são o tipo definidor da convulsão; elas duram até 1 minuto e ocorrem várias vezes por dia.
Também podem ocorrer crises tônico-clônicas, clônicas, atônicas ou de ausência típica.
O EEG ictal mostra descargas de complexos espícula-onda de 3 Hz bilaterais síncronos associados a surtos mioclônicos de 3 Hz; o EEG interictal apresenta uma atividade de base normal, mas até 30% dos pacientes apresentam descargas de complexos espícula-onda generalizados superimpostos.
Os pacientes podem apresentar comprometimento do desenvolvimento na apresentação e alguns posteriormente apresentarão certo grau de deficiência intelectual.
Epilepsia com mioclonia palpebral (EEM):
A idade máxima de início é entre 6 e 8 anos (intervalo de 2 a 14 anos).
A característica mais típica é a mioclonia palpebral que ocorre frequentemente com o fechamento dos olhos, pode ser provocada pela luz (indução fótica) e está frequentemente associada a breves crises de ausência. As convulsões duram apenas alguns segundos, mas ocorrem muitas vezes ao dia.
Outros tipos de convulsão que podem ser observados incluem crises tônico-clônicas generalizadas, mioclônicas e crises de ausência típicas.
EEG: ao fechar os olhos (ou estimulação fótica), são observadas descargas breves de atividades rápidas; um padrão típico são os complexos poliespícula-onda de 3 a 6 Hz.
O desenvolvimento costuma ser normal, mas a deficiência intelectual pode ser observada e é mais provável em pacientes com indução fótica proeminente.
Epilepsia do tipo ausência da infância (EAI):
O início ocorre normalmente entre as idades de 4 e 10 anos (intervalo de 2 a 13 anos).
As crises de ausência típicas são o tipo de convulsão predominante.
Raramente podem ocorrer crises tônico-clônicas generalizadas.
A base do EEG é normal, com paroxismos de espícula-onda generalizados de 3 Hz. O padrão característico da gravação ictal é ictal regular de 3 Hz generalizado e espícula-onda de 3 Hz simétrico.[Figure caption and citation for the preceding image starts]: Eletroencefalograma (EEG): epilepsia do tipo ausência da infância; mostra o típico padrão complexo espícula-onda ritmado a 3 por segundoCortesia do Professor Eric Kossoff; usado com permissão [Citation ends].
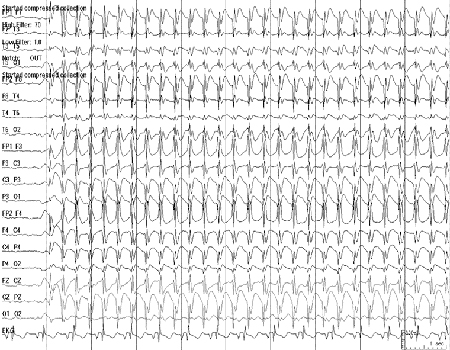
O desenvolvimento é tipicamente normal.
Início em idade variável
O início dessas síndromes geralmente ocorre na terceira infância e na adolescência.[7] O desenvolvimento é normalmente normal. As comorbidades comuns incluem transtornos de humor, TDAH e dificuldades de aprendizagem.
Epilepsia mioclônica juvenil (EMJ):
A idade típica de início é entre 10 e 24 anos (intervalo de 8 a 40 anos).
As crises mioclônicas são obrigatórias para o diagnóstico; podem ser unilaterais ou bilaterais e ocorrem quase exclusivamente pela manhã e principalmente nos membros superiores.
Crises tônico-clônicas generalizadas também ocorrem na maioria dos pacientes e crises de ausência em cerca de um terço dos casos.
O EEG interictal mostra complexos espícula-onda e complexos poliespícula-onda de 3.5 a 6 Hz; mais pronunciados durante o sono e o torpor; espículas rápidas seguidas por ondas lentas irregulares são observadas durante a mioclonia.[Figure caption and citation for the preceding image starts]: Eletroencefalograma (EEG): epilepsia mioclônica juvenilCortesia do Professor Eric Kossoff; usado com permissão [Citation ends].
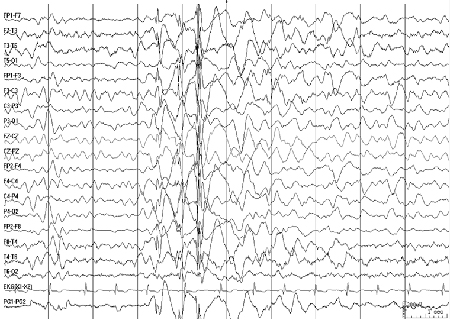
Epilepsia do tipo ausência juvenil (EAJ):
A idade típica de início é entre 9 e 13 anos (intervalo de 8 a 20 anos).
O tipo de convulsão predominante é a crise de ausência. As ausências tendem a ser mais longas e menos frequentes do que as ausências da epilepsia do tipo ausência da infância, e a perda de consciência é muitas vezes menos completa.
Crises tônico-clônicas generalizadas também ocorrem na maioria dos pacientes.
O EEG ictal mostra descargas de complexos espícula-onda lenta generalizados de 3 a 5.5 Hz; o EEG interictal geralmente é normal.
Epilepsia com crises tônico-clônicas generalizadas isoladas (GTCA):
A idade típica de início é entre 10 e 25 anos (intervalo de 5 a 40 anos).
As convulsões tônico-clônicas generalizadas são obrigatórias e nenhum outro tipo de convulsão ocorre. As convulsões geralmente ocorrem 2 horas após acordar e geralmente são pouco frequentes.
O EEG interictal mostra descargas de complexos espícula e/ou poliespícula-onda de 3 a 5.5 Hz.
O uso deste conteúdo está sujeito ao nosso aviso legal