Las manifestaciones principales del déficit de alfa-1-antitripsina (AAT) son pulmonares y hepáticas.
El enfisema panacinar y la enfermedad pulmonar obstructiva asociada son las manifestaciones más comunes.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[38]Marciniuk DD, Hernandez P, Balter M, et al. Alpha-1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clinical practice guideline. Can Respir J. 2012;19:109-116.
https://www.hindawi.com/journals/crj/2012/920918
http://www.ncbi.nlm.nih.gov/pubmed/22536580?tool=bestpractice.com
La evidencia sugiere que casi el 60% de los pacientes desarrolla enfermedades pulmonares graves.[23]Larsson C. Natural history and life expectancy in severe alpha 1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204:345-351.
http://www.ncbi.nlm.nih.gov/pubmed/309708?tool=bestpractice.com
Pueden producirse bronquiectasias. Hasta el 95% de los pacientes con déficit de AAT IP*ZZ tienen evidencia radiológica de bronquiectasias (pero un déficit de AAT IP*ZZ se ha encontrado en <1% de los pacientes que presentan bronquiectasias).[39]Hill AT, Sullivan AL, Chalmers JD, et al. British Thoracic Society Guideline for bronchiectasis in adults. Thorax. 2019 Jan;74(suppl 1):1-69.
https://thorax.bmj.com/content/74/Suppl_1/1.long
http://www.ncbi.nlm.nih.gov/pubmed/30545985?tool=bestpractice.com
En general, la hepatopatía inicialmente se presenta como hepatitis e ictericia, aunque una enfermedad grave puede avanzar hasta convertirse en cirrosis y carcinoma hepatocelular. La afectación del hígado también puede observarse en neonatos con déficit de AAT.[8]Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018 Dec;24 Suppl 1:1-21.
https://www.doi.org/10.1016/j.pulmoe.2018.09.004
http://www.ncbi.nlm.nih.gov/pubmed/30473034?tool=bestpractice.com
[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
La paniculitis necrosante y la granulomatosis con poliangitis son complicaciones poco frecuentes.
La evidencia sugiere que los médicos pueden no reconocer el déficit de AAT como causa de hepatopatía y enfermedad pulmonar.[40]Silverman EK, Miletich JP, Pierce JA, et al. Alpha-1 antitrypsin deficiency: high prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis. 1989;140:961-966.
http://www.ncbi.nlm.nih.gov/pubmed/2679271?tool=bestpractice.com
Hay más evidencia que demuestra una importante diferencia de tiempo entre el inicio de la enfermedad clínica y el diagnóstico, con una evaluación por parte de varios médicos mientras tanto.[41]Stoller JK, Smith P, Yang P, et al. Physical and social impact of alpha-1 antitrypsin deficiency: results of a survey. Cleve Clin J Med. 1994;61:461-467.
http://www.ncbi.nlm.nih.gov/pubmed/7828337?tool=bestpractice.com
Las guías de práctica clínica recomiendan una alta sospecha clínica y una medición cuantitativa de la AAT en los siguientes escenarios:[5]American Thoracic Society/European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha 1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003 Oct 1;168(7):818-900.
https://www.atsjournals.org/doi/full/10.1164/rccm.168.7.818
http://www.ncbi.nlm.nih.gov/pubmed/14522813?tool=bestpractice.com
[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[42]World Health Organization. Alpha-1 antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75:397-415.
http://www.ncbi.nlm.nih.gov/pubmed/9447774?tool=bestpractice.com
[43]Eriksson S, Carlson J, Velez R. Risks for cirrhosis and primary liver cancer in alpha-1 antitrypsin deficiency. N Engl J Med. 1986;314:736-739.
http://www.ncbi.nlm.nih.gov/pubmed/3485248?tool=bestpractice.com
Obstrucción al flujo aéreo parcialmente reversible o irreversible con broncodilatadores.
Todos los pacientes con EPOC[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[44]Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for prevention, diagnosis and management of COPD: 2024 report. 2024 [internet publication].
https://goldcopd.org/2024-gold-report
Todos los pacientes con asma de inicio en la etapa de adulto[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
Antecedentes personales o familiares de vasculitis asociada con anticuerpos anticitoplasma de neutrófilos citoplasmáticos (c-ANCA) (la granulomatosis con poliangeítis es una complicación poco frecuente del déficit de AAT).
Hepatopatía de etiología desconocida.
Bronquiectasias de etiología desconocida, especialmente cuando coexisten con enfisema panacinar[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[39]Hill AT, Sullivan AL, Chalmers JD, et al. British Thoracic Society Guideline for bronchiectasis in adults. Thorax. 2019 Jan;74(suppl 1):1-69.
https://thorax.bmj.com/content/74/Suppl_1/1.long
http://www.ncbi.nlm.nih.gov/pubmed/30545985?tool=bestpractice.com
Paniculitis de etiología desconocida (la paniculitis necrosante es una complicación poco frecuente del déficit de AAT).
Adolescentes y adultos con un hermano que sea homocigoto para AAT.
Personas asintomáticas con disfunción pulmonar obstructiva persistente que reportan tabaquismo o exposición laboral.
Factores históricos
Los síntomas de presentación de las manifestaciones pulmonares son inespecíficos y pueden incluir disnea, disnea de esfuerzo, fatiga, sibilancia, tos u/y opresión en el pecho.
Los síntomas de presentación de las manifestaciones hepáticas son inespecíficos y pueden incluir color amarillo de la piel, fatiga, sangrado, hematomas, distensión abdominal, dolor abdominal y/o confusión.
Es importante considerar la edad, la ocupación y los antecedentes de tabaquismo en pacientes con enfermedad pulmonar sintomática, ya que estos factores pueden indicar un déficit de AAT. El mayor factor de riesgo de enfisema en pacientes con el fenotipo IP*ZZ es el tabaquismo. La función pulmonar y la supervivencia se ven afectadas.[23]Larsson C. Natural history and life expectancy in severe alpha 1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204:345-351.
http://www.ncbi.nlm.nih.gov/pubmed/309708?tool=bestpractice.com
[45]Janus ED, Phillips NT, Carrell RW. Smoking, lung function, and alpha-1-antitrypsin deficiency. Lancet. 1985;1:152-4.
http://www.ncbi.nlm.nih.gov/pubmed/2857224?tool=bestpractice.com
[46]Wu MC, Eriksson S. Lung function, smoking and survival in severe alpha 1-antitrypsin deficiency, PiZZ. J Clin Epidemiol. 1988;41:1157-1165.
http://www.ncbi.nlm.nih.gov/pubmed/3264848?tool=bestpractice.com
No obstante, cierta evidencia sugiere que los ex fumadores y las personas que nunca fumaron presentan un deterioro similar en la función pulmonar con el paso del tiempo, y es posible que algunos fumadores nunca desarrollen síntomas pulmonares.[41]Stoller JK, Smith P, Yang P, et al. Physical and social impact of alpha-1 antitrypsin deficiency: results of a survey. Cleve Clin J Med. 1994;61:461-467.
http://www.ncbi.nlm.nih.gov/pubmed/7828337?tool=bestpractice.com
[47]Piitulainen E, Eriksson S. Decline in FEV1 related to smoking status in individuals with severe alpha-1 antitrypsin deficiency (PiZZ). Eur Respir J. 1999;13:247-251.
http://erj.ersjournals.com/cgi/reprint/13/2/247
http://www.ncbi.nlm.nih.gov/pubmed/10065663?tool=bestpractice.com
La exposición laboral o de otro tipo a gas, emanaciones y/o polvo también se ha asociado con una disminución de la función pulmonar en pacientes con déficit de AAT por IP*ZZ. Esto incluye al tabaquismo pasivo y al trabajo con calentadores de queroseno.[48]Piitulainen E, Tornling G, Eriksson S. Effect of age and occupational exposure to airway irritants on lung function in non-smoking individuals with alpha-1 antitrypsin deficiency (PiZZ). Thorax. 1997;52:244-248.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1758519/pdf/v052p00244.pdf
http://www.ncbi.nlm.nih.gov/pubmed/9093340?tool=bestpractice.com
[49]Piitulainen E, Tornling G, Eriksson S. Environmental correlates of impaired lung function in non-smokers with severe alpha-1 antitrypsin deficiency (PiZZ). Thorax. 1998;53:939-943.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1745103/pdf/v053p00939.pdf
http://www.ncbi.nlm.nih.gov/pubmed/10193391?tool=bestpractice.com
[50]Silverman EK, Pierce JA, Province MA, et al. Variability of pulmonary function in alpha-1 antitrypsin deficiency: clinical correlates. Ann Intern Med. 1989;111:982-991.
http://www.ncbi.nlm.nih.gov/pubmed/2596778?tool=bestpractice.com
[51]Piitulainen E, Sveger T. Effect of environmental and clinical factors on lung function and respiratory symptoms in adolescents with alpha-1 antitrypsin deficiency. Acta Paediatr. 1998;87:1120-1124.
http://www.ncbi.nlm.nih.gov/pubmed/9846912?tool=bestpractice.com
Existe evidencia limitada que sugiere que la enfermedad pulmonar sintomática es más prevalente en los hombres con IP*ZZ que en las mujeres con IP*ZZ. Sin embargo, es probable que este resultado se confunda por otras variables, como el tabaquismo y la exposición laboral.[19]Kueppers F, Fallat R, Larson RK. Obstructive lung disease and alpha-1 antitrypsin deficiency gene heterozygosity. Science. 1969;165:899-901.
http://www.ncbi.nlm.nih.gov/pubmed/5816326?tool=bestpractice.com
[20]Kueppers F, Black LF. Alpha-1 antitrypsin and its deficiency. Am Rev Respir Dis. 1974;110:176-194.
http://www.ncbi.nlm.nih.gov/pubmed/4212922?tool=bestpractice.com
[21]Tobin MJ, Cook PJ, Hutchison DC. Alpha-1 antitrypsin deficiency: the clinical and physiological features of pulmonary emphysema in subjects homozygous for Pi-type-Z. A survey by the British Thoracic Association. Br J Dis Chest. 1983;77:14-27.
http://www.ncbi.nlm.nih.gov/pubmed/6602621?tool=bestpractice.com
[22]Seersholm N, Kok-Jensen A, Dirksen A. Decline in FEV1 among patients with severe hereditary alpha-1 antitrypsin deficiency type Pi Z. Am J Respir Crit Care Med. 1995;152:1922-1925.
http://www.ncbi.nlm.nih.gov/pubmed/8520756?tool=bestpractice.com
La edad media a la que los fumadores con déficit de AAT suelen presentar enfermedades pulmonares sintomáticas es de los 32 a los 41 años.[23]Larsson C. Natural history and life expectancy in severe alpha 1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204:345-351.
http://www.ncbi.nlm.nih.gov/pubmed/309708?tool=bestpractice.com
La historia clínica puede incluir asma o granulomatosis con poliangitis (una complicación poco frecuente del déficit de AAT), y los antecedentes familiares pueden revelar la presencia de déficit de AAT en familiares.
Hallazgos de la exploración
Una inspección general puede revelar ictericia, ictericia conjuntival o asterixis, si hay una hepatopatía presente. La exploración abdominal puede revelar hepatomegalia y/o ascitis.
Un examen respiratorio puede revelar sibilancia y/o tórax distendido, si hay una enfermedad pulmonar presente.
Mediciones séricas de alfa-1-antitripsina (AAT)
Los niveles séricos de AAT se deben cuantificar en personas con posible déficit de AAT.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
Cabe destacar que la AAT también es un reactante de fase aguda, lo que significa que los niveles séricos normales de AAT pueden ser engañosos, especialmente en el entorno de procesos inflamatorios.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
Los estados de la enfermedad pueden estar representados por niveles dudosos o incluso normales de AAT, lo que significa que dichos resultados justifican el mantenimiento de la sospecha. La medición de la AAT en suero por sí sola no se recomienda para las pruebas familiares tras la identificación de un probando porque no caracteriza completamente el riesgo de enfermedad por déficit de AAT.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
Algunas guías de práctica clínica sugieren que el genotipado del alelo S y Z es el primer paso adecuado para las pruebas de diagnóstico de los individuos sintomáticos.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
Pruebas cuantitativas y umbral protector
Niveles bajos a normales de AAT (<35 micromoles/L) deben aumentar la sospecha y motivar la realización de pruebas adicionales. En las pruebas cuantitativas disponibles a nivel comercial, se utilizan métodos de inmunodifusión radial y nefelometría. Los valores umbrales exactos varían según el método de prueba y la orientación regional; deben consultarse las guías de práctica clínica regionales adecuadas para la interpretación de los niveles de AAT en suero.[5]American Thoracic Society/European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha 1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003 Oct 1;168(7):818-900.
https://www.atsjournals.org/doi/full/10.1164/rccm.168.7.818
http://www.ncbi.nlm.nih.gov/pubmed/14522813?tool=bestpractice.com
[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[8]Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018 Dec;24 Suppl 1:1-21.
https://www.doi.org/10.1016/j.pulmoe.2018.09.004
http://www.ncbi.nlm.nih.gov/pubmed/30473034?tool=bestpractice.com
[9]Dummer J, Dobler CC, Holmes M, et al. Diagnosis and treatment of lung disease associated with alpha one-antitrypsin deficiency: a position statement from the Thoracic Society of Australia and New Zealand. Respirology. 2020 Mar;25(3):321-35.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7078913
http://www.ncbi.nlm.nih.gov/pubmed/32030868?tool=bestpractice.com
El nivel de AAT en suero medido con el estándar purificado desarrollado por los Institutos Nacionales de Salud de los Estados Unidos (el método de prueba más común en los Estados Unidos) generalmente se administra en micromol/L, mientras que los niveles de AAT medidos utilizando estándares comerciales generalmente se administran en mg/dL para diferenciarlos.[5]American Thoracic Society/European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha 1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003 Oct 1;168(7):818-900.
https://www.atsjournals.org/doi/full/10.1164/rccm.168.7.818
http://www.ncbi.nlm.nih.gov/pubmed/14522813?tool=bestpractice.com
Los valores de nefelometría menores de 20 micromoles/L (83-120 mg/dL) se consideran deficientes. Se considera que los niveles de nefelometría inferiores a 11 micromol/L (50 mg/dL) confieren una protección inadecuada contra la enfermedad pulmonar inflamatoria; esto se denomina "umbral de protección".[6]Turino GM, Barker AF, Brantly ML, et al. Clinical features of individuals with PI*SZ phenotype of alpha-1 antitrypsin deficiency: alpha 1-antitrypsin deficiency registry study group. Am J Respir Crit Care Med. 1996;154:1718-1725.
http://www.ncbi.nlm.nih.gov/pubmed/8970361?tool=bestpractice.com
Algunos de los fenotipos más comunes dan lugar a los siguientes niveles séricos de AAT:
IP*MM: 20-48 micromoles/L (150-350 mg/dL)
IP*MZ: 17-33 micromoles/L (90-210 mg/dL)
IP*SS: 15-33 micromoles/L (100-200 mg/dL)
IP*ZZ: 2.5-7.0 micromoles/L (20-45 mg/dL).
Aquellos que dan lugar a niveles por debajo del umbral protector tienen mayores probabilidades de causar una enfermedad pulmonar.
Fenotipado (tipaje PI)
Las mediciones por debajo de lo normal de alfa-1-antitripsina (AAT) plasmática pueden corresponder a fenotipos heterocigotos que pueden poner a la persona y a los miembros de su familia en riesgo de sufrir una enfermedad asociada. Los pacientes, y los parientes de primer grado de los pacientes, con niveles normales a bajos pero protectores de AAT (12-35 micromoles/L) deben someterse a pruebas cualitativas a través del fenotipado.
El fenotipado implica la separación de las variantes de AAT usando enfoque isoeléctrico y puede confirmar la identificación de las variantes proteicas de AAT deficientes características. Mediante el fenotipado, se puede revelar la presencia de las variantes proteicas específicas, como la proteína Z, la proteína M (normal) y la proteína S, así como variantes menos comunes.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
Genotipado
Se pueden realizar pruebas genéticas cuando el fenotipo real no corresponda con el fenotipo pronosticado por el nivel sérico de alfa-1-antitripsina (AAT).
Demostrará los alelos característicos de AAT que son responsables de las variantes proteicas de AAT.
Por ejemplo, cuando se detecta un nivel por debajo de lo normal de AAT, se realizan otras pruebas con fenotipado para determinar las variantes proteicas de AAT en el suero. Si solo se detecta la proteína Z, esto no corresponde con un nivel sérico por debajo de lo normal de AAT. En este caso, se podrán realizar pruebas adicionales como el genotipado para determinar los alelos presentes en la persona.
La reacción en cadena de la polimerasa (PCR) se utiliza normalmente para el genotipado. Los alelos poco frecuentes (p. ej., variantes nulas o deficientes distintas de Z o S) pueden requerir una secuencia genética completa. También se puede considerar la secuenciación de genes si no se dispone de cebadores para la PCR.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[9]Dummer J, Dobler CC, Holmes M, et al. Diagnosis and treatment of lung disease associated with alpha one-antitrypsin deficiency: a position statement from the Thoracic Society of Australia and New Zealand. Respirology. 2020 Mar;25(3):321-35.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7078913
http://www.ncbi.nlm.nih.gov/pubmed/32030868?tool=bestpractice.com
Pruebas específicas para las enfermedades respiratorias
Si se presenta una enfermedad respiratoria, las pruebas de función pulmonar demostrarán resultados considerablemente anómalos, incluido un VEF1 reducido.
Mediante radiografías de tórax, se podrán revelar grandes volúmenes pulmonares y enfisema basilar predominante.[Figure caption and citation for the preceding image starts]: Radiografía de tórax de paciente con déficit de AAT (vista posterior-anterior)De la colección personal de D. Kyle Hogarth, MD, FCCP; utilizada con autorización [Citation ends].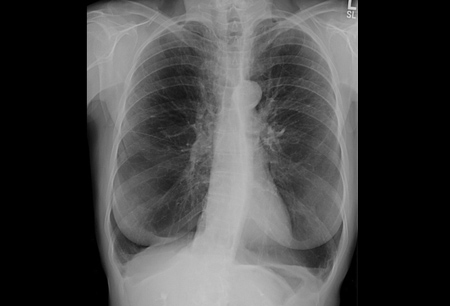 [Figure caption and citation for the preceding image starts]: Radiografía de tórax de paciente con déficit de AAT (vista lateral)De la colección personal de D. Kyle Hogarth, MD, FCCP; utilizada con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Radiografía de tórax de paciente con déficit de AAT (vista lateral)De la colección personal de D. Kyle Hogarth, MD, FCCP; utilizada con autorización [Citation ends].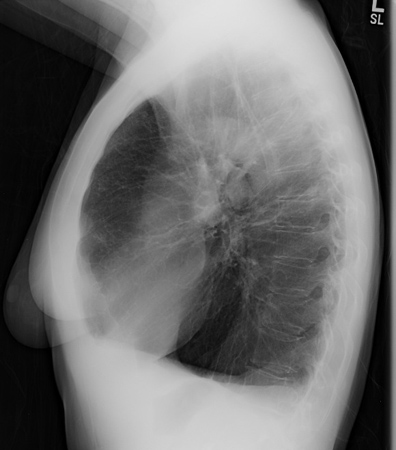
Los pacientes con resultados no diagnósticos pueden requerir una tomografía computarizada (TC) de tórax. La TC es más sensible que las radiografías de tórax o las pruebas funcionales respiratorias para identificar un enfisema panacinar y una bronquiectasia.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[39]Hill AT, Sullivan AL, Chalmers JD, et al. British Thoracic Society Guideline for bronchiectasis in adults. Thorax. 2019 Jan;74(suppl 1):1-69.
https://thorax.bmj.com/content/74/Suppl_1/1.long
http://www.ncbi.nlm.nih.gov/pubmed/30545985?tool=bestpractice.com
Sin embargo, la ausencia de cambios enfisematosos en la TC no descarta un déficit de AAT. El enfisema panacinar se observa principalmente en los lóbulos inferiores, aunque se ha descrito la enfermedad únicamente en los lóbulos superiores. La relación directa entre el déficit de AAT y la bronquiectasia es menos clara, ya que la presencia de bronquiectasia en una TC puede ser el resultado de cambios enfisematosos.[Figure caption and citation for the preceding image starts]: TC de enfisema avanzado en un paciente con déficit de AATDe la colección personal de D. Kyle Hogarth, MD, FCCP; utilizada con autorización [Citation ends].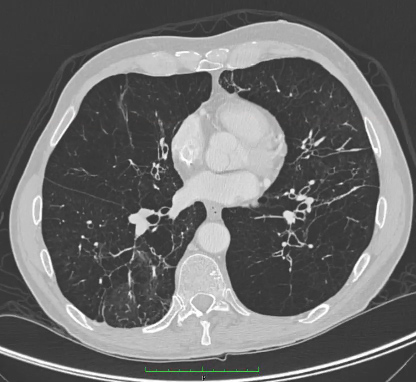
Las pruebas de ejercicio con análisis de gasometría arterial en pacientes con enfisema también suelen ser anómalas y demuestran intolerancia al ejercicio.[7]Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α<sub>1</sub>-antitrypsin deficiency. Eur Respir J. 2017 Nov 30;50(5):1700610.
https://erj.ersjournals.com/content/50/5/1700610.long
http://www.ncbi.nlm.nih.gov/pubmed/29191952?tool=bestpractice.com
[8]Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018 Dec;24 Suppl 1:1-21.
https://www.doi.org/10.1016/j.pulmoe.2018.09.004
http://www.ncbi.nlm.nih.gov/pubmed/30473034?tool=bestpractice.com
Pruebas específicas para enfermedades hepáticas
Las guías de práctica clínica recomiendan la evaluación de la función hepática con pruebas de función hepática (PFH) para aquellos diagnosticados con déficit de AAT, ya sean sintomáticos o asintomáticos para la enfermedad hepática.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
Como parte de cualquier análisis diagnóstico de hepatopatía, los niveles de alfafetoproteína (AFP) también son importantes. No obstante, algunos datos sugieren que la sensibilidad de las PFH, concretamente la alanina aminotransferasa (ALT), es solo del 11.9% para la detección de hepatopatías en el déficit de AAT.[53]Clark VC, Dhanasekaran R, Brantly M, et al. Liver test results do not identify liver disease in adults with alpha-1 antitrypsin deficiency. Clin Gastroenterol Hepatol. 2012;10:1278-1283.
http://www.ncbi.nlm.nih.gov/pubmed/22835581?tool=bestpractice.com
Los pacientes con un fenotipo asociado a la hepatopatía (por ejemplo, IP*ZZ, IP*Mmalton, IP*Siiyama) requieren imágenes hepáticas, y se recomienda una ecografía hepática anual.[37]Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis (Miami). 2016 Jun 6;3(3):668-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5556762
http://www.ncbi.nlm.nih.gov/pubmed/28848891?tool=bestpractice.com
La ecografía hepática también se puede usar para controlar los signos de hipertensión portal y carcinoma hepatocelular. La tomografía computarizada abdominal y/o la resonancia magnética nuclear (RM) también pueden ser útiles para evaluar la morfología hepática, la cirrosis y la hipertensión portal de los pacientes, especialmente en aquellos con obesidad.[52]Guillaud O, Dumortier J, Couchonnal-Bedoya E, et al. Wilson disease and alpha1-antitrypsin deficiency: a review of non-invasive diagnostic tests. Diagnostics (Basel). 2023 Jan 10;13(2):256.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9857715
http://www.ncbi.nlm.nih.gov/pubmed/36673066?tool=bestpractice.com
Si hay carcinoma hepatocelular, las PFH pueden estar empeorando y los niveles de AFP pueden estar aumentando.