O objetivo é confirmar o diagnóstico de DRP autossômica dominante (DRPAD) e então classificar o risco do paciente de evoluir para insuficiência renal com base na combinação de fatores clínicos, genéticos e de imagem.[35]Chebib FT, Torres VE. Recent advances in the management of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2018 Nov 7;13(11):1765-76.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6237066
http://www.ncbi.nlm.nih.gov/pubmed/30049849?tool=bestpractice.com
Manifestações extrarrenais e evidências de complicações de longo prazo de DRPAD também precisam ser verificadas.
Uma história familiar positiva de DRPAD, com sinais e sintomas de manifestações renais e/ou extrarrenais, é altamente sugestiva de DRPAD. O diagnóstico definitivo é obtido a partir de estudos de imagens dos rins ou de testes genéticos, se os exames de imagem forem inconclusivos.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
Um diagnóstico provável pode ser considerado em pacientes sem história familiar positiva. A presença de cistos renais, com ou sem cistos hepáticos, na ausência de outras manifestações é sugestivo de outra doença cística renal.[11]Suwabe T, Shukoor S, Chamberlain AM, et al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol. 2020 Jan 7;15(1):69-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6946081
http://www.ncbi.nlm.nih.gov/pubmed/31791998?tool=bestpractice.com
No entanto, é indicado um teste genético para o diagnóstico definitivo nesses pacientes.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Pelo menos 10% a 15% dos pacientes com história familiar negativa terão uma nova mutação.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Um aconselhamento apropriado deve ser realizado antes do exame (de imagem ou genético) para DRP.[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
A potencial discriminação, em termos de segurabilidade e emprego, associada a um diagnóstico positivo deve ser discutida.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
História
A história familiar pode incluir DRPAD, doença renal em estágio terminal (DRET), aneurisma intracraniano, acidente vascular cerebral (AVC) hemorrágico ou hemorragia subaracnoide. Para adultos com história familiar de DRPAD, o diagnóstico pode ser feito pré-sintomaticamente por imagens abdominais.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Os sintomas manifestos comuns incluem dor abdominal/nos flancos, cólica renal, hematúria macroscópica e, menos comumente, cefaleias.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
[11]Suwabe T, Shukoor S, Chamberlain AM, et al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol. 2020 Jan 7;15(1):69-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6946081
http://www.ncbi.nlm.nih.gov/pubmed/31791998?tool=bestpractice.com
[36]Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008 Oct 2;359(14):1477-85.
http://www.ncbi.nlm.nih.gov/pubmed/18832246?tool=bestpractice.com
As infecções do trato urinário (ITUs) ocorrem em 30% a 50% dos pacientes adultos com DRP.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Pacientes com ITUs do trato inferior apresentam história de disúria, urgência e dor suprapúbica. As infecções do trato urinário (ITUs) envolvendo o parênquima renal ou cistos tipicamente se fazem acompanhar de dor nos flancos e febre. Os pacientes com nefrolitíase podem apresentar dor nos flancos, hematúria, disúria e febre. Pirose, refluxo, náuseas, dispneia, saciedade precoce ou aumento da circunferência abdominal podem ocorrer na doença hepática grave.[37]Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020 Mar 27;12(3):72-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7097502
http://www.ncbi.nlm.nih.gov/pubmed/32231761?tool=bestpractice.com
Há uma associação entre DRPAD e diverticulose na presença de DRET.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
Exame físico
O exame abdominal geralmente revela uma massa renal ou hepática palpável. A DRPAD é caracterizada pelo aumento progressivo de inúmeros cistos renais e do tamanho dos rins, que aumenta exponencialmente com a idade.[26]Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006 May 18;354(20):2122-30.
https://www.nejm.org/doi/10.1056/NEJMoa054341
http://www.ncbi.nlm.nih.gov/pubmed/16707749?tool=bestpractice.com
Os rins aumentam de tamanho e podem estar associados a morbidade significativa devido à massa e peso adicionais.[39]Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006 Jan;1(1):148-57.
https://cjasn.asnjournals.org/content/1/1/148.long
http://www.ncbi.nlm.nih.gov/pubmed/17699202?tool=bestpractice.com
A doença hepática policística está presente em mais de 90% dos indivíduos com DRPAD com mais de 35 anos.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
[37]Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020 Mar 27;12(3):72-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7097502
http://www.ncbi.nlm.nih.gov/pubmed/32231761?tool=bestpractice.com
A hepatomegalia está comumente presente antes mesmo da detecção de cistos hepáticos por imagem.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
A hipertensão é comum e geralmente ocorre em uma idade relativamente jovem, com uma idade média de início entre 30 e 34 anos.[7]Gimpel C, Bergmann C, Bockenhauer D, et al. International consensus statement on the diagnosis and management of autosomal dominant polycystic kidney disease in children and young people. Nat Rev Nephrol. 2019 Nov;15(11):713-26.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7136168
http://www.ncbi.nlm.nih.gov/pubmed/31118499?tool=bestpractice.com
A detecção da hipertensão antes de qualquer outra manifestação clínica é frequentemente o modo como a doença é detectada pela primeira vez em pacientes.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Hérnias inguinais, incisionais ou paraumbilicais frequentemente estão presentes.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
Um sopro cardíaco pode estar presente, sendo sugestivo de prolapso da valva mitral, regurgitação mitral, regurgitação aórtica ou raiz aórtica dilatada.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Exames laboratoriais
Inicialmente, devem ser solicitados exames de eletrólitos, creatinina e ureia séricos e perfil lipídico em jejum. A creatinina pode ser usada para estimar a taxa de filtração glomerular.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
Lipídeos elevados podem estar associados a uma probabilidade maior de insuficiência renal progressiva.[40]Torres VE, Grantham JJ, Chapman AB, et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011 Mar;6(3):640-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3082424
http://www.ncbi.nlm.nih.gov/pubmed/21088290?tool=bestpractice.com
A urinálise deve ser solicitada a todos os pacientes para detectar a presença de aumento na excreção de albumina urinária ou proteinúria. Se for constatado aumento da excreção de albumina urinária ou proteinúria, isso indica uma maior probabilidade de progressão para doença renal crônica.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
[40]Torres VE, Grantham JJ, Chapman AB, et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011 Mar;6(3):640-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3082424
http://www.ncbi.nlm.nih.gov/pubmed/21088290?tool=bestpractice.com
A hematúria microscópica ou macroscópica é comum. Leucocitúria pode ser observada em pessoas com DRP, mas esta nem sempre indica ITU e a cultura de urina deve ser obtida nesse caso. A cultura de urina sempre deve ser obtida na avaliação inicial se houver sintomas de ITU ou febre.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
A cultura de urina pode ser negativa, mesmo com uma infecção urinária grave, porque os cistos não se comunicam com o trato urinário. Em pacientes com dor abdominal ou no flanco e/ou sensibilidade e febre, uma proteína C-reativa deve ser solicitada.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Todos os pacientes com calculose renal metabolicamente ativa devem fazer uma coleta de urina de 24 horas para a bioquímica urinária (volume de urina, pH, oxalatos, ácido úrico, citrato, fosfato, sódio e cálcio, bem como a creatinina para avaliar se foi colhida toda a urina) e deve ser calculada a supersaturação. Citrato e pH urinários baixos são os principais fatores metabólicos que predispõem à formação de cálculo na DRPAD.[41]Torres VE, Erickson SB, Smith LH, et al. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988 Apr;11(4):318-25.
http://www.ncbi.nlm.nih.gov/pubmed/3354568?tool=bestpractice.com
Cintilografia renal
Os exames de imagem exibem a presença de cistos renais com ou sem cistos hepáticos. A ultrassonografia é o exame inicial mais comumente solicitado, devido a custo baixo, ampla disponibilidade e segurança.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
Foram desenvolvidos critérios diagnósticos de ultrassonografia unificados para o diagnóstico de DRPAD em pessoas em risco (aquelas de famílias com DRPAD de genótipo desconhecido).[42]Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009 Jan;20(1):205-12.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2615723
http://www.ncbi.nlm.nih.gov/pubmed/18945943?tool=bestpractice.com
A presença de pelo menos três cistos renais (unilaterais ou bilaterais) e dois cistos em cada rim é suficiente para o diagnóstico de indivíduos de risco com idades entre 15 a 39 e 40 a 59 anos, respectivamente; naqueles com mais de 60 anos, quatro ou mais cistos em cada rim são necessários.[42]Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009 Jan;20(1):205-12.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2615723
http://www.ncbi.nlm.nih.gov/pubmed/18945943?tool=bestpractice.com
Esses critérios não devem ser aplicados a exames de ressonância nuclear magnética (RNM) ou tomografia computadorizada (TC), pois podem levar a resultados falso-positivos.
A TC com contraste ou a RNM detectam cistos de 2 a 3 mm de diâmetro e são particularmente úteis para o diagnóstico em pacientes mais jovens.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
Em pacientes com risco de DRPAD com menos de 30 anos de idade, um critério de teste de um total de >10 cistos renais observados na RNM é considerado suficiente para o diagnóstico.[43]Pei Y, Hwang YH, Conklin J, et al. Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2015 Mar;26(3):746-53.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4341484
http://www.ncbi.nlm.nih.gov/pubmed/25074509?tool=bestpractice.com
O uso da TC precisa ser ponderado em relação à dose de radiação.
A imagem por TC ou RNM deve fazer parte da avaliação inicial da DRPAD, pois fornece medidas precisas e padronizadas do comprimento, largura e profundidade máximos dos rins e uma estimativa do volume total dos rins.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
Esses dados quantitativos são de valor prognóstico.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Um sistema de classificação baseado em imagens no volume renal total da TC ou RNM foi usado para identificar possíveis casos de doença rapidamente progressiva. Este sistema de classificação otimiza a seleção de pacientes para terapia de doenças específicas.[44]Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015 Jan;26(1):160-72.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4279733
http://www.ncbi.nlm.nih.gov/pubmed/24904092?tool=bestpractice.com
Em pacientes com história familiar negativa, o diagnóstico por imagem não é suficiente, pois os critérios foram desenvolvidos em indivíduos com risco de 50% de DRPAD.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
Um provável diagnóstico de DRPAD pode ser considerado se, apesar de não haver história familiar, houver >10 cistos em cada rim e não houver evidência de manifestações renais ou extrarrenais de outra doença renal cística que possa explicar o fenótipo; no entanto, o teste genético é indicado para confirmar o diagnóstico.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
[11]Suwabe T, Shukoor S, Chamberlain AM, et al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol. 2020 Jan 7;15(1):69-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6946081
http://www.ncbi.nlm.nih.gov/pubmed/31791998?tool=bestpractice.com
A tomografia por emissão de pósitrons é mais sensível do que a TC ou a RNM para detectar infecções de cisto.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Tomografia computadorizada (TC) do abdome e da pelve de um paciente com doença leveDa coleção do Dr. M. Hogan [Citation ends].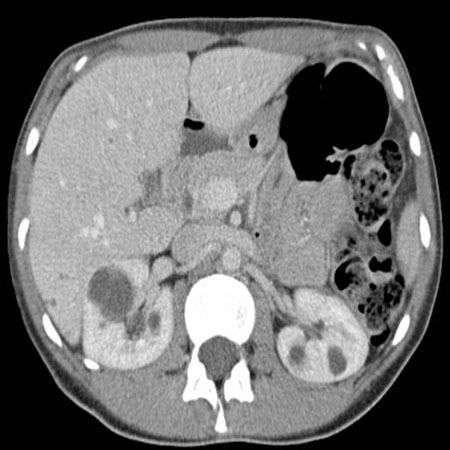 [Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética (RNM) do abdome e da pelve de paciente com doença renal policística sintomáticaDa coleção do Dr. M. Hogan [Citation ends].
[Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética (RNM) do abdome e da pelve de paciente com doença renal policística sintomáticaDa coleção do Dr. M. Hogan [Citation ends].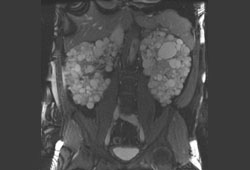
Investigação de manifestações extrarrenais
As anormalidades cardiovasculares incluem hipertensão, hipertrofia ventricular esquerda, dilatação da raiz aórtica, aneurismas arteriais, anormalidades nas valvas cardíacas e aneurismas intracranianos.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
ECG e ecocardiograma são usados para a avaliação inicial de complicações cardiovasculares, como hipertrofia ventricular esquerda (pode estar presente sem hipertensão), função cardíaca (por exemplo, fração de ejeção, disfunção diastólica) e anormalidades valvares.[45]Kuo IY, Chapman AB. Polycystins, ADPKD, and cardiovascular disease. Kidney Int Rep. 2020 Apr;5(4):396-406.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7136326
http://www.ncbi.nlm.nih.gov/pubmed/32274448?tool=bestpractice.com
A TC e a RNM também podem fornecer evidências de cistos extrarrenais. Os cistos hepáticos são as manifestações extrarrenais mais comuns.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Os cistos pancreáticos (prevalência de 9% no rastreamento por ultrassonografia, 36% na RNM) são quase sempre assintomáticos, com raras ocorrências de pancreatite recorrente.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
É raro, porém, possível que estejam associados a tumor mucinoso papilar intraductal ou carcinoma.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
Além disso, se houver suspeita de nefrolitíase, a TC ou a RNM pode diferenciar a calculose renal da calcificação da parede do cisto ou calcificações parenquimatosas. Cerca de 20% dos pacientes têm nefrolitíase e a composição dos cálculos é tipicamente ácido úrico ou oxalato de cálcio.[41]Torres VE, Erickson SB, Smith LH, et al. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988 Apr;11(4):318-25.
http://www.ncbi.nlm.nih.gov/pubmed/3354568?tool=bestpractice.com
[46]Grampsas SA, Chandhoke PS, Fan J, et al. Anatomic and metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000 Jul;36(1):53-7.
http://www.ncbi.nlm.nih.gov/pubmed/10873872?tool=bestpractice.com
A TC de dupla energia é um método útil para diferenciar entre nefrolitíase de ácido úrico e outro tipo de nefrolitíase (não de ácido úrico).[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
A presença de aneurismas intracranianos é quatro vezes maior em pacientes com DRPAD do que na população em geral.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
A identificação de cefaleias sentinelas e o diagnóstico e tratamento imediatos de um aneurisma com vazamento são importantes determinantes do desfecho.[34]Pirson Y, Chauveau D, Torres V. Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2002 Jan;13(1):269-76.
https://jasn.asnjournals.org/content/13/1/269.long
http://www.ncbi.nlm.nih.gov/pubmed/11752048?tool=bestpractice.com
Se houver suspeita de hemorragia subaracnoide, uma TC de crânio sem contraste é realizada, a qual, se não diagnóstica, deve ser seguida por uma punção lombar.[47]Hoh BL, Ko NU, Amin-Hanjani S, et al. 2023 Guideline for the management of patients with aneurysmal subarachnoid hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke. 2023 Jul;54(7):e314-70.
https://www.ahajournals.org/doi/full/10.1161/STR.0000000000000436?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/37212182?tool=bestpractice.com
Se esses testes forem positivos, o paciente deve ser internado imediatamente em uma unidade de terapia intensiva neurocirúrgica para realização de exames de imagem dos vasos, geralmente uma angiotomografia, e tratamento do aneurisma intracraniano em extravasamento.[48]Chung DY, Abdalkader M, Nguyen TN. Aneurysmal subarachnoid hemorrhage. Neurol Clin. 2021 May;39(2):419-42.
http://www.ncbi.nlm.nih.gov/pubmed/33896527?tool=bestpractice.com
Veja nossos tópicos Aneurisma cerebral e Hemorragia subaracnoide.
Teste genético
O teste genético pode ser usado nos seguintes casos:[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
Os resultados dos exames de imagem são duvidosos ou inconclusivos
Para confirmar um diagnóstico presuntivo na ausência de história familiar da doença (a prova conclusiva do diagnóstico nesses pacientes está na análise de mutação)
Quando um diagnóstico definitivo é necessário em um paciente mais jovem, como no caso de um doador de rim vivo com relação de parentesco
Para testes genéticos pré-natais e pré-implantação.
As mutações no gene PKD1 provocam uma doença mais grave que as mutações no gene PKD2.[2]Lavu S, Vaughan LE, Senum SR, et al. The value of genotypic and imaging information to predict functional and structural outcomes in ADPKD. JCI Insight. 2020 Aug 6;5(15):e138724.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7455088
http://www.ncbi.nlm.nih.gov/pubmed/32634120?tool=bestpractice.com
O teste genético para rastreamento da mutação em PKD1 é caro e desafiador devido ao seu grande tamanho e complexidade.[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
Pode ser realizado pela análise de ligação ou de sequência; no entanto, a análise de sequência é preferencial. Apesar do rastreamento abrangente, 10% a 15% dos pacientes com suspeita de DRPAD não têm mutação detectada em PKD1 ou PKD2.[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
Os pacientes podem ser rastreados novamente para mutações de um gene recém-identificado para DRPAD, GANAB, e mosaicismo somático.[20]Iliuta IA, Kalatharan V, Wang K, et al. Polycystic kidney disease without an apparent family history. J Am Soc Nephrol. 2017 Sep;28(9):2768-76.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5576926
http://www.ncbi.nlm.nih.gov/pubmed/28522688?tool=bestpractice.com
Todas as mutações publicadas e outras variantes estão disponíveis ao público no banco de dados de variantes da DRPAD.
PKD Foundation: ADPKD variant database
Opens in new window Se uma mutação for conhecida em um membro familiar, ela pode ser confirmada em outros membros da família com um custo consideravelmente mais baixo.
Uma nova abordagem é o sequenciamento de todo o exoma. Em um estudo de validação, o sequenciamento do genoma completo foi capaz de superar a homologia do pseudogene e identificar todos os tipos de variantes no gene PKD1.[19]Mallawaarachchi AC, Lundie B, Hort Y, et al. Genomic diagnostics in polycystic kidney disease: an assessment of real-world use of whole-genome sequencing. Eur J Hum Genet. 2021 May;29(5):760-70.
http://www.ncbi.nlm.nih.gov/pubmed/33437033?tool=bestpractice.com
