Etiologia
Foram identificados vários genes na doença renal policística autossômica dominante (DRPAD). Os genes mais comumente afetados são PKD1 ou PKD2, com loci menores (por exemplo, GANAB, DNAJB11) sendo responsáveis por uma pequena proporção de pacientes frequentemente atípicos, além de um grupo de casos geneticamente não resolvidos.[2][4][16]
PKD1 (região cromossômica 16p13.3) é encontrado em cerca de 78% dos casos e codifica a proteína policistina 1.[16]
PKD2 (4q21) é encontrado em cerca de 15% dos casos e codifica a policistina 2.[16]
GANAB, um gene codificador que codifica uma subunidade alfa da glucosidase-II, pode ser responsável por aproximadamente 0.3% dos casos de DRPAD.[16] No entanto, devido ao fenótipo leve (rins císticos não aumentados que frequentemente evoluem para atrofia renal) observado em indivíduos com mutações no GANAB, é possível que isso seja responsável por uma proporção maior das causas genéticas ausentes de DRPAD.[17]
Mutações no gene DNAJB11, que codifica uma glicoproteína do retículo endoplasmático (RE) necessária para a homeostase do RE, são uma causa rara (aproximadamente 0.1% dos casos de DRPAD) de DRPAD atípica.[16] Os casos têm rins císticos de tamanho normal e fibrose intersticial progressiva, resultando em doença renal em estágio terminal de início tardio.[18]
A combinação de um alelo da DRPAD com um alelo de outro cistógeno (por exemplo, PKD1 e HNF1B) pode apresentar um fenótipo semelhante ao da DRPAD.[1][19]
Uma história familiar de DRPAD não é aparente em 10% a 25% dos pacientes com DRPAD.[20] Isso pode ser devido à falta de registros médicos dos pais, doença leve de mutações hipomórficas de PKD1 (falha em reconhecer o distúrbio ou doença de início tardio no pai afetado), morte precoce do pai antes do início dos sintomas, mutações de novo ou linha germinativa ou mosaicismo somático.[16][20] Apesar do rastreamento abrangente, 10% a 15% dos pacientes com suspeita de DRPAD não têm mutação detectada em PKD1 ou PKD2.[21] Os pacientes podem ser rastreados novamente para mutações de um gene recém-identificado para DRPAD, GANAB, e mosaicismo somático.[20]
Fisiopatologia
Pacientes com DRP autossômica dominante (DRPAD) geralmente carregam uma mutação das linhas germinativas em um alelo de PKD1 ou PKD2; no entanto, há uma notável variabilidade fenotípica entre os membros da família que compartilham uma mutação comum, sugerindo que uma "segunda incidência" de uma mutação somática do alelo PKD1 previamente normal desempenha um papel importante na determinação do ciclo da doença.[1][22] A teoria é que uma cópia de tipo selvagem do gene PKD1 desenvolve uma mutação somática inativadora em uma minoria de células, levando à perda da função de policistina e ao desenvolvimento de cisto clonal.[22] Os produtos proteicos de PKD1 e PKD2, policistina 1 e policistina 2 são proteínas de membrana que formam um complexo funcional.[23] As evidências apontam para um papel das policistinas no cílio primário do túbulo renal atuando como um canal catiônico mecanossensível, de modo que a perda ou disfunção da policistina 1 ou da policistina 2 altera a concentração de cálcio intracelular, que pode então alterar várias funções celulares, incluindo expressão gênica, crescimento, diferenciação e apoptose, alterando o desenvolvimento de tecidos e órgãos.[24] No entanto, a função biológica das policistinas permanece pouco compreendida.[25]
Os cistos renais se desenvolvem e crescem com o tempo, causando compressão da arquitetura renal normal e da vasculatura intrarrenal com néfrons obstruídos formando glomérulos atubulares e túbulos proximais apoptóticos, tamanho renal aumentado, fibrose intersticial e comprometimento renal progressivo.[1] Os cistos se desenvolvem a partir de células na porção tubular do néfron e dos dutos coletores. Embora todas as células sejam programadas com uma mutação da DRPAD, apenas uma minoria desenvolve cistos. Os rins tornam-se progressivamente maiores e distorcidos, com pouco parênquima reconhecível em estudos de imagem. O aumento do volume renal combinado está associado à diminuição da função renal.[26] A velocidade média de declínio da função renal é de 4.4 a 5.9 mL/minuto/ano.[27][Figure caption and citation for the preceding image starts]: Patologia macroscópica de rins policísticosAdaptado do Dr. Edwin P. Ewing, Jr., Public Health Image Library, CDC (1972) [Citation ends].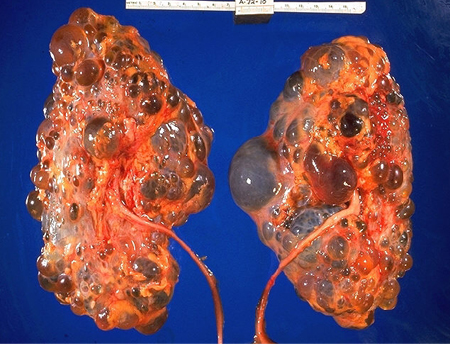
Com base em achados do estudo de referência de Modificação da Dieta na Doença Renal (MDRD - Modification of Diet in Renal Disease), os pacientes com DRPAD apresentaram o declínio mais rápido da função renal entre todas as formas de doença renal crônica.[28] Indivíduos com mutações em PKD1 têm doença mais grave com rins maiores e início precoce de doença renal em estágio terminal (DRET) em comparação com pacientes com mutações em PKD2.[2] A idade média de início da DRET é 55 anos para a mutação em PKD1 e 79 anos para PKD2.[3] Existem menos cistos renais em indivíduos com PKD2 do que naqueles com PKD1, embora a taxa de crescimento dos rins não seja diferente entre os grupos.[29] A DRPAD relacionada a PKD2 é tipicamente mais leve do que a DRPAD relacionada a PKD1; não deve ser considerada assintomática, e o encaminhamento precoce, o cuidado sintomático oportuno e a educação do paciente podem reduzir as modulações ambientais do fenótipo.[30]
Complicações específicas da doença e morbidade adicionais surgem de comprometimento hepático, valvopatia cardíaca, aneurismas intracranianos, dor, aumento maciço dos rins e doença diverticular, contribuindo para morbidade e mortalidade adicionais.[1][31] Os pacientes com PKD1 e PKD2 têm a mesma probabilidade de desenvolver aneurismas intracranianos, embora os pacientes com mutações na região 5' de PKD1 tenham maior probabilidade de ter ruptura do aneurisma.[32]
Na doença renal policística (DRP) autossômica recessiva, os cistos renais surgem a partir dos dutos coletores e a fibrose hepática congênita é característica.[1][33]
Classificação
Doença renal policística autossômica dominante (DRPAD)
A DRPAD é um distúrbio de início típico na idade adulta, caracterizado por cistos renais de crescimento gradual com doença renal fibrocística progressiva.[1] A DRPAD é genética e fenotipicamente heterogênea. Os genes mais comumente afetados são PKD1 ou PKD2.
PKD1 (região cromossômica 16p13.3) codifica a proteína policistina 1.
PKD2 (4q21) codifica a proteína policistina 2.
Indivíduos com mutações em PKD1 têm doença mais grave com rins maiores e início precoce de doença renal em estágio terminal (DRET) em comparação com pacientes com mutações em PKD2.[2] A idade média de início da DRET é 55 anos para a mutação em PKD1 e 79 anos para PKD2.[3]
A DRPAD é uma doença sistêmica e manifestações extrarrenais, como cistos hepáticos e anormalidades cardiovasculares (incluindo aneurismas intracranianos), estão presentes em ambas as variantes de PKD1 e PKD2.[1][4]
Doença renal policística autossômica recessiva (DRPAR)
A DRPAR é uma forma mais rara e frequentemente mais grave de doença cística, que geralmente se apresenta em crianças e fenotipicamente apresenta alta variabilidade, envolvendo os rins e o trato biliar.[1][5] Variantes no gene do rim policístico e doença hepática 1 (PKHD1) são responsáveis pela maioria dos casos de DRPAR, sendo as mutações em DZIP1L uma causa consideravelmente mais rara.[1]
Na sua forma mais grave, ela se apresenta no útero ou no período neonatal com rins ecogênicos aumentados bilateralmente e pode ser de risco de vida.[1][5] Em crianças maiores, ela se manifesta como hipertensão portal ou colangite.[1]
O fenótipo grave de DRPAD de início precoce pode parecer clinicamente indistinguível de DRPAR ou, alternativamente, um ciclo clínico avançado de DRPAR pode se assemelhar aos sintomas observados em DRPAD; entretanto, em contraste com o aumento renal persistente que ocorre na DRPAD, na DRPAR os rins diminuem de tamanho à medida que a quantidade de fibrose aumenta.[1][5] O rastreamento baseado em sequenciamento de próxima geração de um painel de genes-alvo é considerada a abordagem mais eficiente para o diagnóstico.[1]
Este tópico não aborda a DRPAR.
O uso deste conteúdo está sujeito ao nosso aviso legal