Etiologia
A grande maioria dos casos de nefropatia por imunoglobulina A (NIgA) é idiopática, mas ela pode ocorrer de maneira secundária a outras doenças.[8]
A nefropatia por imunoglobulina A hepática, na qual a deposição de IgA mesangial está associada com doença hepática crônica (particularmente cirrose alcoólica), é a forma mais comum de nefropatia por imunoglobulina A secundária.[24][25] Acredita-se que seja uma consequência de uma eliminação hepática deficiente da IgA. A IgA mesangial é um achado de autópsia comum em pacientes com doença hepática crônica, mas apenas uma minoria apresenta manifestações clínicas de doença renal diferente de uma hematúria invisível.
A nefropatia por imunoglobulina A também foi relatada em associação com infecção por vírus da imunodeficiência humana (HIV) e síndrome de imunodeficiência adquirida (AIDS).[26][27][28] O aumento policlonal da IgA sérica, uma característica de AIDS, tem sido citado como um fator predisponente. A força dessa associação é, no entanto, questionável; estudos de autópsia indicaram uma prevalência de nefropatia por imunoglobulina A entre zero e 7.75% em pacientes infectados por HIV. A nefropatia por imunoglobulina A é rara em mieloma por IgA ou outras gamopatias monoclonais por IgA. Essas observações indicam que uma carga circulante alta de IgA monoclonal ou policlonal não é suficiente para promover a deposição por IgA mesangial sem outras anormalidades do sistema IgA.
Evidências indiretas sugerem uma relação próxima entre nefropatia por IgA e a vasculite sistêmica de pequenos vasos por IgA (conhecida anteriormente como púrpura de Henoch-Schönlein [PHS]). A vasculite por IgA é caracterizada por deposição de IgA em vasos sanguíneos de pequeno calibre afetando predominantemente pele, articulações, intestino e rins, com uma nefrite que pode ser histologicamente indistinguível da nefropatia por IgA (NIgA). A nefropatia por IgA é cada vez mais considerada como uma vasculite por IgA sem a erupção cutânea.[29] Não se sabe porque algumas pessoas adquirem doença renal limitada (NIgA), enquanto outras desenvolvem doença sistêmica (vasculite por IgA).
Embora a nefropatia por imunoglobulina A seja comumente esporádica, têm sido relatados casos familiares.[16][17][18][19] O estudo de três novos loci (designados nefropatia por imunoglobulina A1, A2 e A3) não identificou nenhum gene candidato provável. O locus 2q36 identificado em uma linhagem canadense é potencialmente informativo, pois contém os genes COL4A3 e COL4A4, que codificam o colágeno de membrana basal, mutações que estão associadas com nefropatia da membrana basal fina.[30] Quatro estudos de associação genômica ampla da nefropatia por imunoglobulina A demonstraram um enriquecimento de polimorfismos nucleotídeos únicos (PNUs) implicados em traços autoimunes ou inflamatórios (alelos múltiplos dentro da região do antígeno leucocitário humano [HLA] no cromossomo 6p21 e no cromossomo 1q32, sugerindo um papel para proteínas regulatórias complementares).[20][21][22][23] Além disso, um número de loci associados com nefropatia por imunoglobulina A codificam proteínas implicadas na manutenção da barreira intestinal e regulação da resposta imune da mucosa a patógenos.
Fisiopatologia
A patogênese da nefropatia por imunoglobulina A (NIgA) não é completamente compreendida. Foi proposto que o evento desencadeante é a formação de imunocomplexos contendo IgA circulante que são propensos a deposição mesangial. Em pessoas suscetíveis, a IgA depositada gera graus variáveis de inflamação glomerular e a subsequente formação de cicatrizes glomerulares e tubulointersticiais.[31][32] Os imunocomplexos de IgA circulantes são predominantemente compostos de IgA polimérica da subclasse IgA1 de baixa afinidade. A IgA1 presente nesses complexos exibe alterações sutis na composição de carboidratos ligados à região da dobradiça da IgA1. Há uma redução na quantidade de galactose presente nos glicanos na região da dobradiça e sugeriu-se que esta alteração desencadeia a produção de autoanticorpos na nefropatia por imunoglobulina A.[33] Autoanticorpos IgA e imunoglobulina G (IgG) específicos para a região de dobradiça foram detectados no soro de coortes de nefropatia por imunoglobulina A da Europa e Ásia, e acredita-se que promovam formação de imunocomplexo na nefropatia por imunoglobulina A. A função da ativação do complemento na nefropatia por imunoglobulina A ainda não está clara, apesar da identificação frequente de C3 em biópsias renais e a identificação de uma associação entre nefropatia por imunoglobulina A e proteínas reguladoras do complemento em estudos de associação genômica ampla.[34][35]
Classificação
Diversas classificações histopatológicas foram publicadas. A classificação mais amplamente validada entre elas é a classificação de Oxford.[2]
A classificação original de Oxford, publicada em 2009, envolve quatro lesões morfológicas independentes que apresentam reprodutibilidade alta inter- e intraobservador e estão independentemente associadas a um prognóstico desfavorável, independentemente dos achados clínicos ao momento da biópsia renal:
Hipercelularidade mesangial (M0 ou M1)[Figure caption and citation for the preceding image starts]: Hipercelularidade mesangial em nefropatia por IgA (Coloração com ácido periódico de Schiff, x600)Cortesia dos Drs. Hwei Yee Lee, Cristine Ding e Yong Howe Ho (Tan Tock Seng Hospital, Cingapura) [Citation ends].

Hipercelularidade endocapilar (E0 ou E1)[Figure caption and citation for the preceding image starts]: Hipercelularidade endocapilar em nefropatia por IgA (Coloração com ácido periódico de Schiff, x400)Cortesia dos Drs. Hwei Yee Lee, Cristine Ding e Yong Howe Ho (Tan Tock Seng Hospital, Cingapura) [Citation ends].
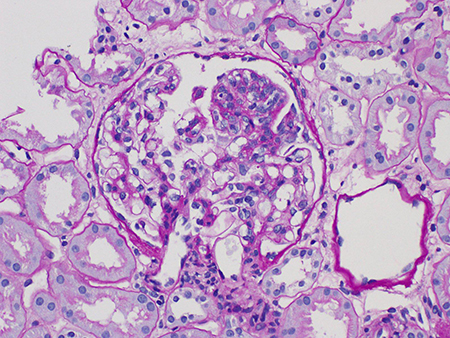
Glomeruloesclerose segmentar (S0 ou S1)[Figure caption and citation for the preceding image starts]: Glomeruloesclerose segmentar em nefropatia por IgA (Coloração com ácido periódico de Schiff, x400)Cortesia dos Drs. Hwei Yee Lee, Cristine Ding e Yong Howe Ho (Tan Tock Seng Hospital, Cingapura) [Citation ends].

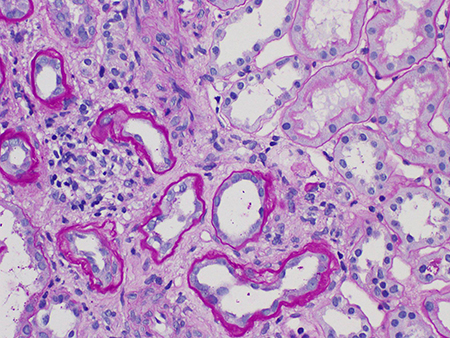
Atrofia tubular/fibrose intersticial (T0, T1 ou T2).[Figure caption and citation for the preceding image starts]: Atrofia tubular e fibrose intersticial em nefropatia por IgA (os túbulos atrofiados são vistos à esquerda do campo, e os túbulos normais são vistos à direita) (Coloração com ácido periódico de Schiff, x400)Cortesia dos Drs. Hwei Yee Lee, Cristine Ding e Yong Howe Ho (Tan Tock Seng Hospital, Cingapura) [Citation ends].
Desde sua publicação, a classificação de Oxford foi validada em diversas coortes de pacientes da América do Norte, Europa e Ásia.[3][4][5]
Na Classificação atualizada de Oxford, publicada em 2017, crescentes foram adicionadas às quatro lesões originais: C0 (sem crescentes), C1 (crescentes em menos de 25% dos glomérulos) e C2 (crescentes em 25% ou mais dos glomérulos), formando um novo escore MEST-C.[6] Esse é o resultado de um subgrupo de trabalho do Classification Working Group que demonstrou que crescentes (C1 ou C2) eram preditores independentes de desfechos renais em uma coorte agrupada de 3096 pacientes.[7] Outra adição notável à Classificação atualizada de Oxford de 2017 é a subclassificação de esclerose segmentar, identificando aqueles casos com evidências de hipertrofia podocitária e/ou lesão de ponta.
Ainda que a definição patológica de nefropatia por IgA seja simples, as variações dos fenótipos são marcantes: estão incluídas variações quanto a incidência geográfica, quadro clínico e progressão, histopatologia renal e recorrência de transplante.
Não há atualmente sistemas de classificação clínica para pacientes com nefropatia por imunoglobulina A que reflitam precisamente o fenótipo variado desta glomerulonefrite complexa.
Atualmente, não há prova de que a entidade conhecida como nefropatia por imunoglobulina A seja uma "doença" única (usando o termo "doença" em seu sentido convencional de uma única entidade compartilhando fatores etiológicos e processos patogênicos). Também não há provas de que a nefropatia por imunoglobulina A é a mesma "doença" em todas as partes do mundo.
O uso deste conteúdo está sujeito ao nosso aviso legal