Abordagem
Uma história pessoal característica e familiar, combinada a achados do exame físico, aumenta a suspeita para o diagnóstico de hemofilia congênita. Isso é confirmado, subsequentemente, por achados laboratoriais. A idade da apresentação e a frequência do sangramento são influenciados pela gravidade do quadro clínico. A maioria dos pacientes é diagnosticada quando criança. No entanto, alguns pacientes podem permanecer não diagnosticados até a idade adulta, particularmente aqueles com hemofilia leve ou mesmo moderada cujo sistema hemostático não tenha sido submetido a testes de desafio significativos durante a vida. As hemofilias A e B são clinicamente indistinguíveis.
A hemofilia adquirida é uma doença autoimune rara resultante da produção de autoanticorpos que inativam o fator VIII. Muitas vezes, não é reconhecida ou é confundida com outras doenças hemorrágicas adquiridas, o que resulta em diagnóstico e manejo tardios.
História
Os fatores de risco fortemente associados à hemofilia congênita incluem:
História familiar de hemofilia, geralmente positiva do lado materno (tios, primos, avô)
Sexo masculino.
O sangramento musculoesquelético é o traço característico da hemofilia. Uma história típica nos pacientes com hemofilia congênita inclui sintomas de sangramento intenso ou recorrente, ou sangramento em articulações ou músculos. São comuns relatos de sangramento mucocutâneo menor (por exemplo, epistaxe, sangramento gengival após procedimentos odontológicos simples, hematomas frequentes) e de sangramento intenso após um trauma, cirurgia ou procedimento odontológico.
Sangramento gastrointestinal e hematúria são observados em pacientes com hemofilia congênita. Eles podem ocorrer espontaneamente ou após trauma. Mulheres e meninas portadoras de hemofilia congênita, principalmente aquelas com níveis de fator de coagulação na faixa de hemofilia, muito comumente apresentam menorragia e sangramento após procedimentos cirúrgicos ou parto.[25]
No período neonatal, as apresentações a seguir são as mais comuns de hemofilia congênita:
Sangramento intracraniano que pode ocorrer espontaneamente após o nascimento, ou pode ser precipitado por trabalho de parto prolongado ou parto instrumental
Sangramento prolongado após picada no calcanhar
Sangramento prolongado após circuncisão: o sangramento ocorre em cerca de 50% dos neonatos com hemofilia submetidos à circuncisão.[26]
Aproximadamente metade dos casos de hemofilia adquirida está associada a doenças autoimunes, doença maligna, determinados medicamentos ou gestação. Ao contrário dos pacientes com hemofilia congênita, esses pacientes não têm história pessoal nem familiar de episódios de sangramento.
Exame físico
Os achados típicos do exame físico de pacientes com hemofilia congênita variam dependendo da idade do paciente, da gravidade do tipo de hemofilia e do local do sangramento. Cerca de 3% a 5% dos neonatos do sexo masculino com hemofilia congênita grave apresentam hemorragia intracraniana.[27][28][29][30] Os sinais e sintomas são inespecíficos, mas incluem hipoatividade, ingestão oral reduzida, irritabilidade, fontanela abaulada/tensa, convulsões e palidez.
Outros possíveis sinais na apresentação neonatal incluem:
Sangramento ativo em um local de lesão
Dor/edema em um dos membros
Abdome distendido e doloroso.
Os sinais típicos de apresentação na infância ou na fase adulta incluem:
Sangramento ativo em um local de lesão
Dor/edema nas articulações
Dor/edema de membros
Amplitude de movimento reduzida de um membro
Abdome distendido e doloroso
Palidez
Deficits neurológicos focais
Hematúria.
Caso ocorra hemorragia intracraniana na infância, sinais e sintomas podem incluir hipoatividade, irritabilidade, cefaleia, vômitos, convulsões e deficits neurológicos focais. Embora o deficit de crescimento e o retardo do crescimento pôndero-estatural possam ser atribuídos à presença de hemofilia, eles não são sinais comuns; portanto, deve-se buscar um diagnóstico alternativo.
O sangramento musculoesquelético é o traço característico da hemofilia congênita. Locais comuns de sangramento incluem articulações do joelho, tornozelo e cotovelo, embora possa haver o envolvimento de qualquer articulação. A apresentação usual inclui dor, edema e amplitude de movimento reduzida nas articulações. O sangramento muscular pode ocorrer em qualquer músculo, incluindo, mas não limitado aos seguintes:
Quadríceps
Isquiossurais
Iliopsoas
Bíceps
Tríceps.
A apresentação usual do sangramento muscular inclui dor localizada, edema, aumento da temperatura, eritema e amplitude de movimento limitada. Nos pacientes com hemofilia grave (que não seguem medidas de profilaxia), o sangramento musculoesquelético pode ocorrer até uma vez por semana; Nos pacientes com hemofilia moderada, o sangramento musculoesquelético pode ocorrer uma vez ao mês ou menos, e ele pode ocorrer cerca de uma vez ao ano ou menos naqueles com hemofilia leve, e geralmente apenas com trauma significativo ou procedimento invasivo/cirurgia.[31][32][33]
Formação de hematomas ou contusões excessivas também podem ocorrer. Os membros inferiores são os mais afetados, embora outros locais possam ser acometidos. O sangramento no músculo iliopsoas pode apresentar dor intensa na parte inferior do abdome, na parte superior da coxa e/ou dor na coluna lombar. Os pacientes sentem dor com a extensão, mas não com a rotação, da articulação do quadril e, normalmente, apresentam uma marcha característica (quadril flexionado, rotação interna).
Em pacientes com a forma adquirida, os achados do exame físico incluem púrpura cutânea extensa e sinais de hemorragia interna. O sangramento nas articulações não é um traço característico proeminente. A razão para o padrão de sangramento diferenciado na hemofilia adquirida é desconhecida. Não há comprometimento demonstrável da função plaquetária.[Figure caption and citation for the preceding image starts]: Hemartrose aguda do joelho direito com equimoseDepartamento de Hematologia Pediátrica, University of Texas Health Science Center, Houston; usada com permissão [Citation ends].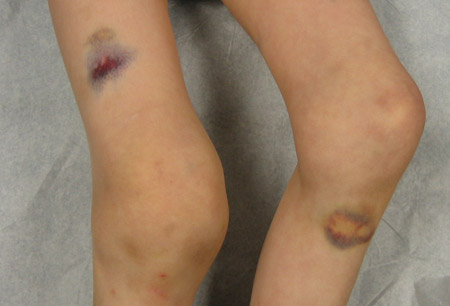 [Figure caption and citation for the preceding image starts]: Hemartroses bilaterais agudas dos joelhosDepartamento de Hematologia Pediátrica, University of Texas Health Science Center, Houston; usada com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Hemartroses bilaterais agudas dos joelhosDepartamento de Hematologia Pediátrica, University of Texas Health Science Center, Houston; usada com permissão [Citation ends].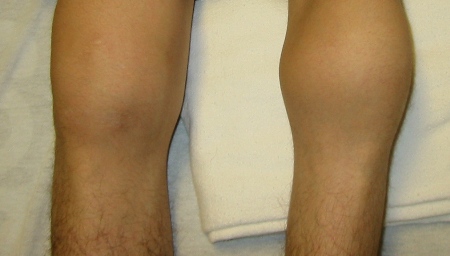 [Figure caption and citation for the preceding image starts]: Edema maciço causado por hemartrose aguda do joelho direitoDepartamento de Hematologia Pediátrica, University of Texas Health Science Center, Houston; usada com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Edema maciço causado por hemartrose aguda do joelho direitoDepartamento de Hematologia Pediátrica, University of Texas Health Science Center, Houston; usada com permissão [Citation ends].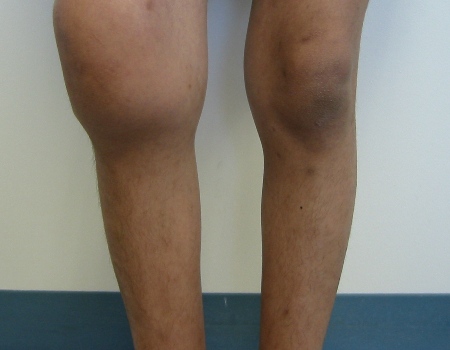
Investigações laboratoriais iniciais
O tempo de tromboplastina parcial ativada (TTPa) deve ser solicitado se houver suspeita de um diagnóstico de hemofilia congênita. Caso o TTPa seja prolongado, deve-se solicitar um ensaio do fator VIII e/ou do fator IX para confirmar o diagnóstico. O TTPa nem sempre está prolongado nos casos leves (níveis de fator >30%). Entretanto, se o diagnóstico for clinicamente suspeito, recomenda-se solicitar ensaios dos fatores VIII e IX. Se o TTPa estiver prolongado, convém também solicitar um estudo misto (incubando o plasma do paciente com plasma normal por 2 horas a 37 °C [98.6°F] e com a repetição do TTPa). A correção com estudos de mistura sugere uma deficiência de fator de coagulação, enquanto a falta de correção sugere a presença de um inibidor de coagulação.
O diagnóstico de hemofilia adquirida baseia-se no achado de um baixo nível de fator VIII associado à presença de um inibidor tempo-dependente no plasma. Na hemofilia adquirida, o TTPa prolongado não pode ser corrigido pela incubação do plasma do paciente com plasma normal por 2 horas a 37 °C (98.6°F) devido à presença de inibidor do fator VIII dependente de tempo e temperatura
Outros exames indicados como parte da investigação diferencial incluem:
Hemograma completo: para descartar trombocitopenia como causa de sangramento e para diagnosticar anemia
Tempo de protrombina (TP): para avaliar as vias extrínseca e comum da coagulação
Tempo de fechamento/tempo de sangramento e estudos de agregação plaquetária: para avaliar a função plaquetária
Estudos do fator de von Willebrand: para descartar a doença de von Willebrand
Outros ensaios específicos para o fator: conforme necessário, baseados nos resultados de TP e TTPa (por exemplo, se o TP estiver prolongado, deve-se verificar os ensaios do fator VII e fator V para descartar uma deficiência coexistente do fator VII ou o raro estado de deficiência autossômica recessiva combinada de fator V e VIII; se apenas o TTPa estiver prolongado, mas os fatores VIII e IX estiverem normais, deve-se verificar também os ensaios dos fatores XII e XI)
Aminotransferases hepáticas (aspartato transaminase [AST] e alanina aminotransferase [ALT]): para avaliar a disfunção hepática, que também pode contribuir para TP e TTPa prolongados
Estudo misto e teste de rastreamento de inibidor de lúpus para descartar TTPa prolongado mediado pelo inibidor.
Exames de imagem e endoscopia
Estudos de imagem ou endoscopia podem ser necessários para avaliação de sangramento agudo. As investigações, conforme adequado, podem incluir:
Tomografia computadorizada (TC) e/ou ressonância nuclear magnética (RNM) de crânio: para avaliação de hemorragia intracraniana[30]
Tomografia computadorizada (TC) e/ou ressonância nuclear magnética (RNM) de crânio: para avaliação de sangramento próximo às vias aéreas
Ultrassonografia abdominal ou TC abdominopélvica: para avaliação de hemorragia digestiva ou sangramento do iliopsoas
Endoscopia digestiva alta e/ou baixa: para avaliação de hemorragia digestiva
Radiografia simples: conforme necessário, para avaliação óssea. As radiografias são tradicionalmente usadas para descrever a progressão clínica da artropatia.[34] A RNM e a ultrassonografia podem detectar sangramentos de tecidos moles em estágio inicial. A RNM pode ser útil para avaliar se um paciente é candidato a procedimentos cirúrgicos, como sinovectomia, artroplastia ou fusão de articulação.
Investigações subsequentes
Pode incluir análise de mutações, teste de rastreamento para inibidor de fator VIII ou IX e ensaio de inibidor de Bethesda. Essas investigações são reservadas para os pacientes com diagnóstico de hemofilia.
Análise da mutação
A análise de mutação dos fatores VIII ou IX identifica mutações genéticas específicas envolvidas na hemofilia A e B. Ela é realizada para estabelecer:
Precisão diagnóstica
Gravidade clínica
Risco de desenvolvimento de inibidor.
A análise de mutação é recomendada para os pacientes com diagnóstico de hemofilia A ou B, parentes do sexo masculino afetados e parentes do sexo feminino com risco de serem portadoras.[35] Também pode ser usada para fundamentar o diagnóstico pré-natal.
A análise de mutação deve ser realizada em centros especializados, com experiência em testes genéticos para hemofilia. O aconselhamento genético deve ser fornecido antes e depois da testagem.[35]
Os pacientes portadores de hemofilia B com mutações genéticas resultantes de deleções requerem observação cuidadosa, pois podem desenvolver anafilaxia e/ou síndrome nefrótica quando o fator IX for infundido.[36][37] Geralmente, uma reação alérgica ao fator IX indica que o paciente está desenvolvendo ou desenvolveu um inibidor do fator IX.
Teste de rastreamento de inibidor de fator VIII ou IX
O rastreamento detecta a presença de anticorpos inibitórios contra os fatores VIII ou IX infundidos. As indicações para rastreamento de inibidor incluem:[38]
Exposição inicial ao fator
Exposição intensa ao fator (por exemplo, exposição diária por mais de 5 dias)
Sangramentos recorrentes ou sangramentos em uma articulação-alvo (apesar da terapia de reposição adequada do concentrado de fator de coagulação)
Fracasso na resposta à terapia de reposição adequada do concentrado de fator de coagulação
Recuperação do fator ou meia-vida abaixo do esperado após a terapia de reposição de concentrado do fator de coagulação
Resposta clínica ou laboratorial abaixo do ideal à terapia de reposição de concentrado do fator de coagulação
Antes da cirurgia
Resposta pós-operatória abaixo do ideal à terapia de reposição de concentrado do fator de coagulação.
Os pacientes com hemofilia leve que não recebem infusões com frequência podem ser monitorados a cada 12 meses. Se os pacientes com inibidores apresentarem sangramento agudo, o tratamento será complexo e deverá ser manejado em centros especializados.
Cerca de 30% dos pacientes com hemofilia A desenvolvem inibidores contra o fator VIII infundido, enquanto a incidência de inibidor para hemofilia B é de até 5%.[38]
Os fatores de risco para o desenvolvimento de inibidores incluem:[19][22][39][40][41][42][43][44][45][46][47][48]
Gravidade da doença: mais comum na hemofilia grave (mas também pode ocorrer em pacientes com hemofilia leve a moderada)
Fatores ambientais: intensidade do tratamento, infecção, idade, tipo de produto
Mutações genéticas: em particular, as que resultam na ausência de um produto gênico; genes da resposta imune também estão associados ao desenvolvimento de inibidores. As pessoas com história familiar de desenvolvimento de inibidores contra o fator VIII infundido e pessoas com ascendência afrocaribenha ou hispânica apresentam maior probabilidade de desenvolverem inibidores.
Nenhuma evidência foi encontrada para dar suporte a uma associação entre o desenvolvimento de inibidores e a imunização ou o momento da imunização relativa à infusão do fator VIII.[49][50]
A possibilidade de haver uma diferença quanto à incidência de inibidores entre receptores de produtos de fator VIII recombinante e derivados do plasma está sob intensa investigação. Alguns estudos não revelaram nenhuma diferença, enquanto um estudo de pacientes com hemofilia A grave que não receberam transfusão previamente sugeriu que a incidência de inibidores foi mais elevada entre os pacientes que tinham recebido o fator VIII recombinante que naqueles randomizados para receber o fator VIII derivado de plasma.[51][52][53][54] A National Hemophilia Foundation dos EUA orienta que o desenvolvimento de inibidores é um risco significativo no tratamento dos pacientes com hemofilia A previamente não tratada, e que os inibidores do fator VIII podem ocorrer com concentrados de fator VIII recombinantes e derivados de plasma.[54]
Ensaio de inibidor de Bethesda
Caso o rastreamento de anticorpos seja positivo, um ensaio de inibidor de Bethesda (ou um dos ensaios modificados com sensibilidade aprimorada, como o ensaio de inibidor de Bethesda modificado por Nijmegen ou o ensaio de inibidor de Bethesda-Nijmegen modificado pelos Centros de Controle e Prevenção de Doenças [CDC] é realizado para medir a quantidade de anticorpos inibitórios presente na amostra do paciente (em unidades Bethesda [UB]).[55][56] O ensaio é realizado misturando-se o plasma do paciente a uma quantidade conhecida de fator VIII ou IX. Após um período de incubação de 2 horas a 37 °C (98.6 °F), determina-se a atividade residual do fator VIII ou IX. Os pacientes são classificados como de baixa ou alta resposta com base nos níveis de anticorpos inibitórios, e essa classificação contribui para a tomada de decisão sobre o tratamento.[1] Os pacientes com alta resposta apresentam história de título ≥5 UB; entretanto, convém observar que os pacientes com alta resposta podem apresentar um resultado de baixo título (um título <5 UB) em qualquer teste específico sem que altere a classificação geral.
Os inibidores também podem ser quantificados usando o método de ensaio de inibidor de Bethesda do substrato cromogênico, que tem a vantagem de reduzir a falsa positividade.[57]
Diagnóstico pré-natal
Mulheres que sejam sabidamente portadoras, com risco de dar à luz uma criança com hemofilia congênita, podem buscar um diagnóstico pré-natal.[58] A presença de ácido desoxirribonucleico (DNA) do feto no sangue materno pode ser detectada até 4 a 5 semanas de gestação, o que permite a determinação do sexo masculino. O sexo também pode ser determinado por ultrassonografia no início da 11ª a 13ª semana de gestação. A análise do cromossomo X baseada no DNA para identificar uma mutação genética específica pode ser realizada na amniocentese ou na biópsia da vilosidade coriônica (BVC). A amniocentese geralmente é realizada após a 15ª semana de gestação, enquanto a BVC pode ser realizada entre a 10ª e a 14ª semana. Uma anormalidade nos membros foi associada à BVC realizada antes de 10 semanas de gestação, e a mãe deve ser orientada sobre o pequeno risco de aborto espontâneo com BVC e amniocentese. Se a mutação for conhecida, poderá ser realizada uma análise de mutação direta. Se a mutação ainda não for conhecida, poderá ser realizado o sequenciamento do gene para determinar a presença de mutação. O diagnóstico genético pré-implantacional em embriões precoces também é possível.[38][59]
O uso deste conteúdo está sujeito ao nosso aviso legal