Abordagem
Existem várias características típicas da doença de Charcot-Marie-Tooth (CMT).[8] Os indivíduos afetados geralmente têm dificuldades com o equilíbrio e fraqueza nos tornozelos desde a infância. Os reflexos tendinosos profundos estão geralmente ausentes ou reduzidos difusamente tanto nos membros inferiores quanto nos superiores, embora o sítio mais constante seja o tendão de Aquiles. Os estudos de condução nervosa mostrarão uma redução na velocidade, com amplitudes reduzidas e latências distais prolongadas na desmielinização na CMT tipo 1 (CMT1), ou amplitudes reduzidas com velocidades normais na presença de perda axonal na CMT tipo 2 (CMT2).
A presença de uma neuropatia periférica com uma história familiar positiva e/ou teste genético anormal tem valor diagnóstico. Quando não há história familiar de CMT e o teste genético para identificar a doença resulta negativo, é frequentemente difícil distinguir entre CMT e uma neuropatia inflamatória adquirida. Nessas situações, pode-se usar uma biópsia do nervo sural para identificar a presença de outros distúrbios em vez de CMT.
A gravidade da CMT pode ser determinada usando-se o Escore de Neuropatia para Charcot-Marie-Tooth (CMTNS), uma métrica validada de deterioração para pessoas com a doença.[9] O CMTNS foi atualizado em 2011, dando origem ao CMTNSv2, que diminuiu os efeitos-teto do CMTNS.[10] Em uma escala de 36 pontos que inclui os sintomas, achados clínicos e eletrofisiologia, uma pontuação de 0-10 representa danos leves, de 11-20, danos moderados, e ≥21, danos graves. Um subconjunto dessa escala, chamado Escore de Exames para CMT (CMT Examination Score; CMTES), foi atualizado em 2020 usando a análise de Rasch e pode fornecer uma análise rápida de como um indivíduo está clinicamente, usando os sinais e sintomas do CMTNS, mas não os escores eletrofisiológicos.[11]
História clínica
Para determinar se uma pessoa tem CMT, é importante realizar uma anamnese completa, obtendo as histórias familiar, médica e de desenvolvimento detalhadas.
Características da apresentação
As características mais comuns são fraqueza e atrofia da perna e do pé.[Figure caption and citation for the preceding image starts]: Pés cavos, pododáctilos em martelo e atrofia muscular peroneal bilateral em paciente com doença de Charcot-Marie-Tooth tipo 1A (CMT1A)Adaptado de Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
 A maioria dos indivíduos tem dificuldades para andar, torce os tornozelos e bate os pés contra o chão ao caminhar. Os nervos do músculo tibial anterior são preferencialmente afetados, causando fraqueza dos tornozelos e perda da habilidade da dorsiflexão do pé.[8] As características são normalmente simétricas, embora assimetrias possam acontecer ocasionalmente.
A maioria dos indivíduos tem dificuldades para andar, torce os tornozelos e bate os pés contra o chão ao caminhar. Os nervos do músculo tibial anterior são preferencialmente afetados, causando fraqueza dos tornozelos e perda da habilidade da dorsiflexão do pé.[8] As características são normalmente simétricas, embora assimetrias possam acontecer ocasionalmente.Os sintomas acontecem de forma comprimento-dependente, já que a maioria dos nervos distais se degenera (perda axonal) primeiro. Assim, os músculos mais distais são afetados primeiro, sendo os pés afetados antes dos tornozelos que, por sua vez, são afetados antes das mãos.[8] Sensações anormais, como perda de sensibilidade e queimação ou parestesia nas mãos e nos pés, geralmente começam nos pododáctilos e prosseguem em direção proximal ao longo do tempo.[Figure caption and citation for the preceding image starts]: Perda de massa muscular das mãos em paciente com doença de Charcot-Marie-Tooth tipo 1A (CMT1A)Adaptado de Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
 [Figure caption and citation for the preceding image starts]: Pés cavos, pododáctilos em martelo e atrofia muscular peroneal bilateral em paciente com doença de Charcot-Marie-Tooth tipo 1A (CMT1A)Adaptado de Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
[Figure caption and citation for the preceding image starts]: Pés cavos, pododáctilos em martelo e atrofia muscular peroneal bilateral em paciente com doença de Charcot-Marie-Tooth tipo 1A (CMT1A)Adaptado de Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].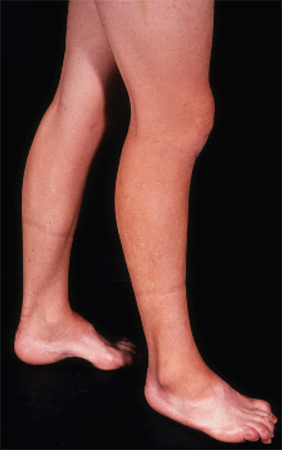
Crianças com marcos motores tardios, que nunca correram ou deambularam, têm um fenótipo grave de início precoce da CMT1, CMT2 e CMT4, chamado síndrome de Dejerine-Sottas. A neuropatia hereditária com predisposição a paralisia por pressão (NHPP) se apresenta com sintomas sensoriais e motores temporários após uma compressão leve ou o alongamento de um nervo. Esses sintomas podem durar de semanas a meses e são geralmente assimétricos. A maioria das pessoas com CMT tipo 2A (CMT2A) tem fraqueza motora significativa nos anos pré-escolares e escolares e até os 20 anos terá perdido a capacidade de deambular, embora o comprometimento sensorial só seja afetado posteriormente na evolução da doença. Esses indivíduos podem também apresentar fraqueza muscular proximal. A curvatura da coluna (cifoescoliose) pode ocorrer em algumas pessoas com CMT e é uma característica proeminente em algumas formas (por exemplo, CMT4C devida a mutações em SH3TC2).
História médica pregressa
As questões principais sobre a história do desenvolvimento incluem dificuldades com o equilíbrio ou patinação na infância e problemas em achar sapatos confortáveis devido aos pés com arcos elevados.
Deve-se também observar a ocorrência de procedimentos cirúrgicos prévios nos pés e tornozelos. Transferências de tendão nos pés e alongamento dos tendões de Aquiles são cirurgias comuns realizadas por causa de tendões de Aquiles rígidos. Outras cirurgias incluem o realinhamento dos pododáctilos em martelo e a tríplice artrodese do tornozelo.
História familiar
Como a CMT é uma neuropatia periférica hereditária, é importante obter a história familiar de, pelo menos, três gerações. A maioria dos indivíduos afetados tem história familiar da doença. Uma história familiar de neuropatia, pés cavos (arcos dos pés elevados com pododáctilos em martelo) ou marcha irregular é um indício forte de doença hereditária, especialmente de CMT.
É importante desenhar uma árvore genealógica e investigar ambos os lados da família por três gerações para determinar quem é afetado e qual é o padrão de herança presente. Deve-se fazer perguntas como “esse parente tinha/tem algum problema para andar, de equilíbrio, tropeços, quedas ou com as mãos ou sensações?”. Uma linhagem autossômica dominante terá pessoas afetadas em todas as gerações, e uma transmissão homem-a-homem pode estar presente. Linhagens ligadas ao cromossomo X não terão transmissão homem-a-homem, e os homens serão quase sempre afetados mais gravemente que as mulheres. Histórias autossômicas recessivas podem ter somente uma pessoa ou irmão afetado.
A falta de história familiar não exclui o diagnóstico, já que mutações de novo são relativamente comuns.[12][13] Há também variabilidades fenotípicas em que os pais ou outros membros da família podem não estar cientes de suas doenças, e casos recessivos podem estar presentes sem história familiar.
Exame físico
O exame neurológico deve testar os nervos cranianos, força, sensação, reflexos, coordenação e deambulação.
O exame dos nervos cranianos geralmente é normal. Alguns indivíduos com CMT tipo 1B (CMT1B) apresentam anormalidades de constrição pupilar, e as pessoas com CMT2A podem apresentar anormalidades nos nervos ópticos.
Ataxia sensorial (isto é, desequilíbrio e falta de coordenação devido à perda de propriocepção) pode estar presente.
Força reduzida nos músculos distais dos braços e pernas (por exemplo, musculatura intrínseca dos pés e das mãos, tibial anterior) com atrofia associada e enfraquecimento da eversão do pé. Essa observação clínica indica uma doença do neurônio motor inferior comprimento-dependente. A força é geralmente mantida nos músculos proximais e no músculo gastrocnêmio.[8][Figure caption and citation for the preceding image starts]: Perda de massa muscular das mãos em paciente com doença de Charcot-Marie-Tooth tipo 1A (CMT1A)Adaptado de Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
[Figure caption and citation for the preceding image starts]: Pés cavos, pododáctilos em martelo e atrofia muscular peroneal bilateral em paciente com doença de Charcot-Marie-Tooth tipo 1A (CMT1A)Adaptado de Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].A sensação que corresponde tanto às fibras nervosas sensoriais grandes quanto pequenas é diminuída ou ausente.[8] Há uma redução na sensibilidade à dor e na sensação de vibração, que é mais acentuada nos pododáctilos que nas regiões proximais.[8]
Os reflexos tendinosos profundos estão difusamente ausentes (arreflexia) ou reduzidos (hiporreflexia).[8] A perda de reflexos pode ocorrer em qualquer doença que envolva danos aos nervos, mas a natureza difusa dos locais afetados não é típica de outras doenças.
Pés com arcos elevados e pododáctilos em martelo (pés cavos) são comuns, mas não patognomônicos, já que os pés cavos podem ser observados em uma variedade de doenças e podem estar presentes sem um problema neurológico. Se os pés cavos forem associados à arreflexia, a probabilidade de CMT é alta. Os pés cavos resultam de um desequilíbrio muscular, tendo o tibial anterior e a musculatura intrínseca dos pés afetados, mas não o músculo gastrocnêmio. A tração mais forte do gastrocnêmio supera a tração mais fraca do tibial anterior, causando as deformidades estruturais do pé.[8][Figure caption and citation for the preceding image starts]: Pés cavos, pododáctilos em martelo e atrofia muscular peroneal bilateral em paciente com doença de Charcot-Marie-Tooth tipo 1A (CMT1A)Adaptado de Berciano J, Gallardo E, Garcia A, et al. Charcot-Marie-Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 2006 Oct;77(10):1169-76 [Citation ends].
A avaliação da marcha revela batida dos pés contra o chão, ou perda do padrão calcanhar-pododáctilos, podendo a pessoa afetada caminhar em marcha em "steppage" (levantamento excessivo das pernas para liberar os pododáctilos). A marcha digitígrada e a ausência de marcha sobre os calcanhares resultam de um tendão de Aquiles rígido devido ao comprometimento preferencial dos nervos do tibial anterior, ocasionando força insuficiente para levantar os pés durante a deambulação.[8] Os pacientes podem solicitar suportes bilaterais, como órteses tornozelo-pé, para deambular.
Exames por imagem
Uma radiografia do quadril deve ser considerada em todas as crianças para rastrear a displasia do quadril.[14] Radiografias da coluna cervical, torácica e lombar e da pelve (série espinhal) devem ser consideradas nas crianças que apresentem sinais de escoliose ou cifose. Recomenda-se obter as incidências posteroanterior e lateral.
Teste da função pulmonar
O teste da função pulmonar basal deve ser realizado nas seguintes crianças quando elas forem capazes de completá-lo de forma confiável (geralmente a partir dos 5-6 anos de idade):[14]
Crianças com sintomas de distúrbios respiratórios do sono (por exemplo, cefaleias inexplicáveis, sonolência diurna ou sintomas de apneia obstrutiva do sono).
Crianças com infecções recorrentes do trato respiratório inferior (>2 ciclos de antibióticos durante 4 meses ou >2 internações hospitalares por infecção do trato respiratório em 12 meses).
Crianças com escoliose (ângulo de Cobb >40°).
Todas as crianças que não deambulam.
Estudos de condução nervosa (ECN)
Deve-se considerar essa investigação para qualquer indivíduo com suspeita de neuropatia, pois ela pode confirmar o diagnóstico de uma polineuropatia e determinar se a doença é essencialmente motora, sensorial ou mista, e se afeta a mielina e a célula de Schwann ou o axônio. Devem-se estudar tanto os nervos sensoriais quanto os motores, embora valha ressaltar que a interpretação dos resultados pode ser difícil, já que algumas formas de CMT apresentam apenas anormalidades leves nos ECN.
As anormalidades dos nervos são simétricas, com um grau similar de desaceleração ou redução de amplitude em todos os nervos testados, e as respostas dos nervos sensoriais estão geralmente ausentes. A desmielinização na CMT1 (inclusive a CMT tipo 1A [CMT1A]) é associada a velocidades de condução muito baixas, de 15 a 38 m/segundo, respostas sensoriais ausentes ou reduzidas e latências distais prolongadas. A perda axonal na CMT2 (inclusive a CMT2A) é associada a amplitudes reduzidas (especialmente, na CMT2A), mas com velocidades de condução relativamente normais, >38 m/segundo.
Na presença de atrofia extrema nos músculos distais, as respostas podem não ser identificáveis e pode ser necessário realizar os ECN em um músculo mais proximal que o usual.
Teste genético
Há mais de 90 causas genéticas conhecidas, e nos Estados Unidos há testes comerciais disponíveis para a maioria delas.
Deve-se realizar o teste genético somente após um aconselhamento adequado. As justificativas para se determinar a etiologia genética específica da doença incluem o conforto psicológico, o planejamento familiar, a habilidade de participar de ensaios clínicos e o fornecimento de mais conhecimento sobre a história natural do subtipo particular de CMT. Essas considerações devem ser ponderadas com a possibilidade de discriminação no trabalho e em seguros, o custo para o indivíduo, o estigma associado a um diagnóstico genético e a dinâmica familiar.
Uma abordagem lógica baseada no teste de condução nervosa e no histórico genético pode otimizar o teste genético.[7][15] Embora se saiba que mais de 90 genes possam ocasionar a CMT quando mutados, cerca de 92% das pessoas com uma mutação identificável terão uma mutação em 1 de 4 genes: PMP22 (duplicação ou deleção), GJB1, MPZ ou MFN2.[16] Os fenótipos podem ajudar a definir o subtipo de CMT, usando-se a porção do nervo afetada (mielina ou axônio), a forma de herança (autossômica dominante, autossômica recessiva ou ligada ao cromossomo X) e a idade de início (primeira infância, infância, fase adulta).[7] Além disso, a testagem genética pode ser otimizado com base nos números da prevalência. As pessoas com uma árvore genealógica que mostre uma mutação dominante ou transmissão homem-a-homem e sinais de desmielinização nos ECN devem ser testadas para CMT tipo 1A (CMT1A), a forma mais comum da CMT desmielinizante. Se não houver duplicação do gene PMP22 indicativa de CMT1A, pode ser adequado testar outras doenças genéticas que afetem a mielina. Se houver possibilidade de uma doença ligada ao cromossomo X, é adequado testar para CMT1X com mutações do gene GJB1. Uma mutação causativa do gene é identificada em aproximadamente 80% dos casos de CMT desmielinizante.[17] As pessoas com um padrão axonal nos ECN, com ou sem história familiar, devem ser testadas para mutações no MFN2 que causem CMT2A, a forma mais comum de CMT axonal.[13] Apenas 30% a 40% dos casos de CMT axonal apresentarão uma mutação identificável.
Os painéis de sequenciamento de nova geração possibilitam opções rápidas e baratas para diversos genes de uma só vez. Estudos de condução nervosa, história familiar e exame físico podem ajudar na interpretação dos resultados de perfis extensos. Por exemplo, se uma variante for encontrada em um gene axonal, mas o indivíduo tiver CMT desmielinizante, é improvável que o gene axonal variante seja o causador da doença. O sequenciamento do exoma está rapidamente se tornando mais econômico e útil em encontrar novos genes na CMT. Se uma pessoa tiver o teste genético negativo para as causas mais comuns da CMT, pode ser recomendável encaminhar o ácido desoxirribonucleico (DNA) da pessoa e de outros membros da família afetados para pesquisa ou análise clínica do exoma.
Os relatórios de resultados devem ser interpretados com cuidado. A detecção de uma mutação genética conhecida, associada a uma forma de CMT hereditária dominante, é definitivamente a causa da doença. Entretanto, uma única mutação causadora de doença em um gene recessivo não pode ser a causa. As variações genéticas de significância incerta são geralmente mutações que alteram aminoácidos e que ainda não foram classificadas como causadoras de doenças ou benignas, geralmente só podendo ser interpretadas ao testar os pais e rastrear para verificar se a doença e a variante segregam-se juntas. É sempre apropriado ligar para o laboratório que realizou o teste para esclarecer e interpretar os resultados.
Recomenda-se consultar um especialista em uma clínica que atenda várias pessoas com CMT para fazer os testes mais eficientes e precisos. Muitos especialistas primeiro traçariam um perfil e, depois, fariam um sequenciamento do exoma, caso necessário.
O uso deste conteúdo está sujeito ao nosso aviso legal