Abordagem
A avaliação inicial de pacientes com suspeita de trombocitemia essencial (TE) inclui anamnese e exame físico para avaliar sinais e sintomas de trombose e possíveis causas de trombocitose.
O diagnóstico de TE é baseado na classificação da Organização Mundial da Saúde e requer avaliação de características clínicas e laboratoriais (sangue e medula óssea) e exclusão de causas secundárias ou reativas de trombocitose (consulte Critérios de diagnóstico).[2]
Uma vez estabelecido o diagnóstico de TE, é importante estabelecer o risco de cada paciente evoluir para sangramento ou complicações trombóticas. Isso possibilitará a definição dos tratamentos terapêuticos e preventivos mais apropriados.
História
Deve-se fazer uma anamnese completa, inclusive com história de cirurgia recente, trauma, neoplasia, infecção, esplenectomia prévia e presença de anemia ferropriva. Esses fatores podem causar trombocitose reativa ou secundária e precisam ser descartados.
Fatores de risco para complicações trombóticas ou hemorrágicas
Uma história de certos fatores de risco irá determinar se os pacientes apresentam risco baixo ou elevado de complicações trombóticas ou hemorrágicas. Esses fatores de risco incluem presença de comorbidade trombótica ou hemorrágica, bem como fatores de risco cardiovascular, incluindo história de cardiopatia isquêmica, hipertensão, diabetes mellitus, acidente vascular cerebral (AVC) ou trombofilia familiar.
Em gestantes, é importante determinar história de gestações prévias, abortos espontâneos, sangramento e perda fetal. Entre gestantes, a TE está associada a aumento do risco de aborto espontâneo.[10]
Sinais e sintomas
Aproximadamente 40% a 50% dos pacientes com TE são assintomáticos no momento do diagnóstico, sendo a trombocitose um achado incidental em exames de sangue de rotina.[3][4]
Os pacientes sintomáticos costumam apresentar sintomas vasomotores ou complicações provenientes da trombose ou sangramento.[3][5]
Os sintomas vasomotores podem incluir cefaleia, tontura, dor torácica, parestesia, vertigem e eritromelalgia (caracterizada por dor com queimação e congestão escura dos membros). Síncope, convulsões e distúrbios visuais transitórios são manifestações vasomotoras incomuns. Os sintomas vasomotores não são específicos da TE e, independentemente da causa, uma alta contagem plaquetária pode estar associada a esses sintomas.
Os eventos trombóticos podem incluir AVC, ataques isquêmicos transitórios (AITs), oclusões da artéria retiniana ou venosas, oclusão da artéria coronária, embolia pulmonar, trombose da veia porta ou hepática, trombose venosa profunda, isquemia digital e priapismo (uma complicação rara relacionada à trombose do corpo cavernoso).[6] A isquemia digital pode se manifestar inicialmente como fenômeno de Raynaud, com palidez e/ou cianose dos dígitos, mas pode progredir para necrose isquêmica das falanges terminais. Pacientes com AIT devem ser submetidos a ultrassonografia das carótidas para descartar estenose de artéria carótida.
Geralmente, os eventos hemorrágicos são leves e se manifestam como epistaxe ou facilidade em apresentar hematomas. O trato gastrointestinal é o local mais comum para sangramento importante.[7] O aumento do risco de sangramento está associado à trombocitose extrema (contagem plaquetária >1000 x 10⁹/L [>1 milhão/microlitro]) e ao uso de aspirina em doses >325 mg/dia.[35]
Exame físico
Não existem sinais específicos de TE, mas a ausência de condições que podem causar trombocitose secundária ou reativa, juntamente com a presença de esplenomegalia, pode sugerir o diagnóstico. A esplenomegalia está presente em aproximadamente 10% a 20% dos pacientes com TE no momento do diagnóstico e, geralmente, tem grau modesto.[8][9]
Os pacientes devem ser examinados em busca de sinais de anemia, infecção, neoplasia ou uma cicatriz cirúrgica condizente com esplenectomia para descartar causas secundárias de trombocitose.
O livedo reticular (uma descoloração arroxeada e mosqueada na pele, geralmente nas pernas; tipicamente descrita como tendo aparência rendada ou reticulada) pode ocorrer na TE, mas é também observado em várias doenças do tecido conjuntivo (por exemplo, lúpus, síndrome antifosfolipídica ou síndrome de Sneddon). Isso pode ajudar a estabelecer o diagnóstico de trombocitose reativa.
Exames laboratoriais iniciais
Os exames laboratoriais iniciais incluem hemograma completo (com diferencial), esfregaço de sangue periférico e perfil de ferro sérico. Achados laboratoriais podem ajudar a descartar possíveis causas de trombocitose reativa ou secundária (por exemplo, condições inflamatórias ou infecciosas, esplenectomia e deficiência de ferro).
Hemograma completo
Pacientes com TE apresentarão contagem plaquetária elevada, persistente e inexplicável. A trombocitose é a principal característica da TE.
A contagem plaquetária pode variar de 450 × 10⁹/L a >1000 × 10⁹/L (450,000 a >1 milhão/microlitro). O grau de trombocitose não pode ser usado para predizer a probabilidade de TE; a contagem plaquetária pode estar elevada aos mesmos níveis observados em pessoas com trombocitose reativa. Se a trombocitose for relatada no estudo inicial, ela deve ser confirmada pela repetição de exames e análise de esfregaço de sangue periférico.[36]
A contagem de leucócitos é geralmente normal, mas pode estar discretamente elevada na TE. Pacientes com trombocitose reativa causada por doenças inflamatórias ou infecciosas podem apresentar uma contagem de leucócitos elevada.
Esfregaço de sangue periférico
Em pacientes com TE, o esfregaço de sangue periférico mostrará trombocitose com vários graus de anisocitose plaquetária (variando de plaquetas com tamanho normal e granulação normal a plaquetas grandes [trombócitos] hipogranulares). Células precursoras imaturas (por exemplo, mielócitos, metamielócitos) podem também ser observadas.
Eritrócitos no esfregaço de sangue periférico são geralmente normocrômicos e normocíticos em pacientes com TE. Eritrócitos hipocrômicos e microcíticos podem indicar deficiência de ferro (uma causa de trombocitose reativa).
Pacientes submetidos a esplenectomia ou com redução da função esplênica (hipoesplenismo) podem apresentar trombocitose secundária. O esfregaço de sangue periférico nesses pacientes mostrará fragmentos nucleares (corpos de Howell-Jolly) em eritrócitos, juntamente com células-alvo e eritrócitos disformes.
Perfis séricos do ferro
O baixo nível de ferritina sérica (por exemplo, <27 picomoles/L [<12 nanogramas/mL]; varia entre diretrizes) é diagnóstico de deficiência de ferro, com especificidade próxima a 100%.
Exames subsequentes
Os testes para causas específicas de trombocitose devem se basear na suspeita de diagnóstico observado na avaliação clínica.
Testes de proteína C-reativa sérica, VHS e fibrinogênio
A proteína C-reativa sérica (PCR), a velocidade de hemossedimentação (VHS) e o fibrinogênio são reagentes de fase aguda e marcadores de inflamação subjacente. Geralmente, são normais na TE e elevados na maioria dos casos de trombocitose reativa.
Exame da medula óssea
É indicado se houver evidências de trombocitose no hemograma completo e no esfregaço de sangue periférico sem uma causa identificável. A biópsia da medula óssea em pacientes com TE mostrará proliferação da linhagem de megacariócitos com aumento do número de megacariócitos maduros e aumentados com núcleos hiperlobulados. Além disso, não haverá aumento significativo ou desvio à esquerda na granulopoiese de neutrófilos ou eritropoiese, e nenhum aumento importante nas fibras de reticulina. Um aumento menor (grau 1) nas fibras de reticulina pode ser observado, mas é raro. Um aumento importante nas fibras de reticulina sugere uma mielofibrose primária pré-fibrótica/precoce (prePMF). Distinguir entre TE e prePMF pode ser um desafio.[37] É fundamental fazer essa distinção na biópsia da medula óssea, pois esses dois distúrbios têm prognósticos significativamente diferentes.[Figure caption and citation for the preceding image starts]: Achados na medula óssea característicos de trombocitemia essencial em biópsias por trefina coradas com hematoxilina e eosina. Aumento na celularidade (seta grossa) e aumento no número e tamanho de megacariócitos maduros com núcleos hiperlobulados (seta fina)Reproduzido com a permissão de Michiels JJ, de Raeve H, Hebeda K, et al. Leuk Res. 2007 Aug;31(8):1031-8 [Citation ends].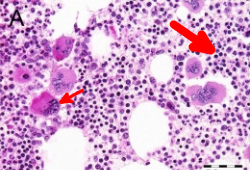
Teste de mutação genética (JAK2 V617F, CALR e MPL)
Todos os pacientes com suspeita de TE devem ser submetidos inicialmente a testes moleculares (no sangue periférico ou, alternativamente, na medula óssea) para a mutação Janus quinase 2 (JAK2) V617F. Se negativo, devem ser realizados testes para mutações no gene da calreticulina (CALR) e no oncogene do vírus da leucemia mieloproliferativa (MPL).[38] De forma alternativa, um painel de sequenciamento de nova geração abrangendo todas as três mutações condutoras de neoplasia mieloproliferativa (NMP) pode ser usado.
A presença de mutação JAK2 V617F, em CALR ou em MPL indica uma NMP, mas essas mutações condutoras não são específicas para TE. As mutações JAK2 V617F, em CALR e em MPL estão presentes em aproximadamente 50% a 60%, 25% a 30% e 3% a 11% dos pacientes com TE, respectivamente.[19] Aproximadamente 10% a 15% dos pacientes com TE são negativos para todas as três mutações condutoras (triplo negativos); portanto, a ausência dessas mutações não descarta o diagnóstico.[19] As mutações JAK2 V617F, em CALR e em MPL podem ocorrer em outras neoplasias mieloproliferativas (policitemia vera, mielofibrose primária) e neoplasias malignas mieloides.
A expressão das mutações condutoras é frequentemente considerada mutuamente exclusiva na TE; entretanto, há relatos de pacientes com mutações coexistentes de JAK2 V617F e CALR ou de JAK2 V617F e MPL.[20][21]
O tratamento da TE pode ser individualizado com base no estado da mutação
Mutação JAK2 V617F: associada a maior risco de trombose (particularmente entre homozigotos [>50% de carga alélica mutante] e aqueles que são mais jovens [idade <60 anos]) e um menor risco de mielofibrose pós-TE.[27][28][29][30][31][39] Pacientes com a mutação JAK2 V617F exibem maior contagem leucocitária e níveis de hemoglobina do que aqueles sem a mutação.[29][32]
A mutação em CALR está associada a menor idade, sexo masculino, nível mais baixo de hemoglobina, maior contagem plaquetária, menor contagem de leucócitos e menor risco de trombose, em comparação com a TE com mutação JAK2 V617F.[39][40]
Mutação em MPL: inconsistentemente associada a idade avançada, sexo feminino, nível de hemoglobina mais baixo, maior contagem plaquetária e possivelmente menor sobrevida livre de mielofibrose, em comparação com a TE com MPL do tipo selvagem.[39]
Testes citogenéticos e moleculares: BCR::ABL1
Deve-se realizar uma hibridização in situ fluorescente (FISH) ou reação em cadeia da polimerase para detectar BCR::ABL1 (cromossomo Filadélfia).[38]
A ausência de BCR::ABL1 ajuda a descartar leucemia mieloide crônica (LMC), uma neoplasia mieloproliferativa que pode se manifestar, inicialmente, com trombocitose isolada. É importante descartar a LMC, pois o prognóstico e o tratamento dessa doença são muito diferentes daqueles da TE.[41]
O uso deste conteúdo está sujeito ao nosso aviso legal