Etiologia
A causa da trombocitemia essencial (TE) é desconhecida.
Fatores genéticos podem estar envolvidos no desenvolvimento da TE. As mutações condutoras somáticas nos genes de Janus quinase 2 (JAK2), calreticulina (CALR) ou oncogene do vírus da leucemia mieloproliferativa (MPL) estão comumente presentes em pacientes com TE e outras neoplasias mieloproliferativas (mielofibrose primária, policitemia vera).[19] Essas mutações condutoras podem fazer com que células sanguíneas anormais cresçam e se proliferem incontrolavelmente.
Mutações de JAK2 V617F, CALR e MPL estão presentes em aproximadamente 50% a 60%, 25% a 30% e 3% a 11% dos pacientes com TE, respectivamente.[19] Aproximadamente 10% a 15% dos pacientes com TE são negativos para todas as três mutações condutoras (triplo negativos); portanto, a ausência dessas mutações não descarta o diagnóstico.[19] A expressão das mutações condutoras é frequentemente considerada mutuamente exclusiva na TE; entretanto, há relatos de pacientes com mutações coexistentes de JAK2 V617F e CALR ou de JAK2 V617F e MPL.[20][21]
Há relatos de TE familiar com transmissão autossômica dominante, mas mutações específicas da linha germinativa que causam TE familiar não foram identificadas.[22][23][24]
Como em muitas neoplasias hematológicas, fatores ambientais e de estilo de vida podem ser contribuintes.[25] No entanto, atualmente não existem circunstâncias ou agentes etiológicos claramente identificados.
Fisiopatologia
A TE é um distúrbio clonal de células-tronco hematopoiéticas. Caracteriza-se pela proliferação de megacariócitos anormais na medula óssea, resultando em aumento da produção de plaquetas, o que causa trombocitose. A causa e o processo fisiopatológico que causa o aumento da produção de plaquetas não são claros.
Mutações genéticas da trombopoetina (TPO) não têm sido implicadas na patogênese da TE. No entanto, em um estudo sobre uma família com trombocitemia hereditária, mutações nos genes da trombopoetina ou c-MPL foram associadas à trombocitose.[26]
O mecanismo pelo qual a TE produz trombose ou sangramento ainda não foi bem definido. Vários defeitos têm sido descritos, inclusive hiperagregação e aumento da ativação plaquetária (que causa trombose) e diminuição da agregação (que causa sangramento).
A mutação de JAK2 V617F está associada ao aumento da ativação de plaquetas e leucócitos e a um estado hipercoagulável, particularmente entre homozigotos (>50% da carga de alelos mutantes) e pessoas mais jovens (<60 anos de idade).[27][28][29][30][31] Pacientes com a mutação JAK2 V617F exibem maiores níveis de hemoglobina do que aqueles sem a mutação.[29][32]
Foram também relatados decréscimos na atividade do cofator de ristocetina de von Willebrand, multímeros de fator de von Willebrand de alto peso molecular, antitrombina III, proteína C e proteína S.[33][34] Há maior probabilidade de sangramento em pacientes com trombocitose extrema (ou seja, contagem plaquetária >1500 x 10⁹/L [>1.5 milhão/microlitro]) em decorrência de uma deficiência adquirida de fator de von Willebrand.[Figure caption and citation for the preceding image starts]: Achados na medula óssea característicos de trombocitemia essencial em biópsias por trefina coradas com hematoxilina e eosina. Aumento na celularidade (seta grossa) e aumento no número e tamanho de megacariócitos maduros com núcleos hiperlobulados (seta fina)Reproduzido com a permissão de Michiels JJ, de Raeve H, Hebeda K, et al. Leuk Res. 2007 Aug;31(8):1031-8 [Citation ends].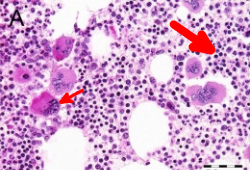
Classificação
O uso deste conteúdo está sujeito ao nosso aviso legal