Etiologia
O gene da glicose-6-fosfato desidrogenase (G6PD) está localizado na extremidade do braço longo do cromossomo X. Portanto, a deficiência é observada em homens hemizigotos (que têm uma mutação em seu único cromossomo X) e nas mulheres homozigotas (que têm uma mutação em cada um de seus cromossomos X; muito raramente observada na maioria das doenças ligadas ao cromossomo X, mas frequente em populações em que a deficiência de G6PD é comum [por exemplo, na África]).
A alta prevalência de mutações na G6PD pode ser explicada pelo benefício protetor que os alelos deficientes em G6PD conferem contra o parasita da malária Plasmodium falciparum (semelhante à observada na doença falciforme e nas talassemias). A natureza exata do mecanismo de proteção é incerta. As evidências que apoiam isso são derivadas de estudos populacionais que mostram que a distribuição global de alelos deficientes em G6PD é semelhante à distribuição mundial da malária.[4] Além disso, um estudo clínico mostrou que a presença de mutações específicas (por exemplo, no alelo da G6PD A) reduz o risco de malária grave por Plasmodium falciparum em 46% a 58%.[9] Estudos in vitro mostraram que, em mulheres heterozigotas, a parasitação dos eritrócitos normais por esse micro-organismo é 2 a 80 vezes maior que nas células deficientes em G6PD.[4]
As mulheres heterozigotas são mosaicos genéticos: metade de suas células é normal; a outra metade é completamente deficiente em G6PD. Elas em geral são apenas levemente sintomáticas, mas às vezes ocorre hemólise aguda grave. As mutações no gene causam substituições de aminoácidos (ou, raramente, pequenas deleções) que alteram a estrutura tridimensional da enzima e causam a atividade enzimática reduzida. Aproximadamente 160 mutações foram identificadas, muito menos que o número de variantes bioquímicas previamente descritas. Muitas dessas variantes parecem ter surgido independentemente em diferentes partes do mundo; agora, sabe-se que são idênticas ao nível molecular.[10] As relações de função da estrutura foram definidas com a correlação clínica. Portanto, as mutações subjacentes às variantes da classe I resultam em enzimas muito mais instáveis e são associadas a uma atividade acentuadamente reduzida, enquanto as mutações que causam as variantes da classe III e IV são associadas a uma conservação maior da atividade enzimática.[3][11]
As variantes genéticas específicas mais comuns incluem:
G6PD mediterrânea: uma variante de classe II que afeta as pessoas de países europeus e árabes próximos ao Mediterrâneo (ou seus descendentes)
G6PD A-: uma variante de classe III que afeta as pessoas da África Subsaariana (ou seus descendentes)
G6PD Mahidol (classe III) e Union (II): comumente afetam populações asiáticas.
Fisiopatologia
A G6PD é necessária para todas as células, a fim de protegê-las dos danos por oxidação. Ela catalisa a primeira etapa na via de pentose-fosfato, na qual a glicose-6-fosfato é oxidada para 6-fosfogliconato. Essa reação está ligada à redução de NADP (nicotinamida-adenina dinucleotídeo fosfato) a NADPH (nicotinamida-adenina dinucleotídeo fosfato reduzida), que é usado para gerar a glutationa reduzida.
[Figure caption and citation for the preceding image starts]: Função da glicose-6-fosfato desidrogenase na via de pentose-fosfato, resultando na geração de nicotinamida-adenina dinucleotídeo fosfato reduzida (NADPH) e glutationa reduzida (GSH), produtos necessários para proteger o eritrócito do estresse oxidativoAdaptado de Haematology at a Glance, Mehta AB and Hoffbrand AV, 3rd Edition Wiley Blackwell 2007; usado com permissão [Citation ends].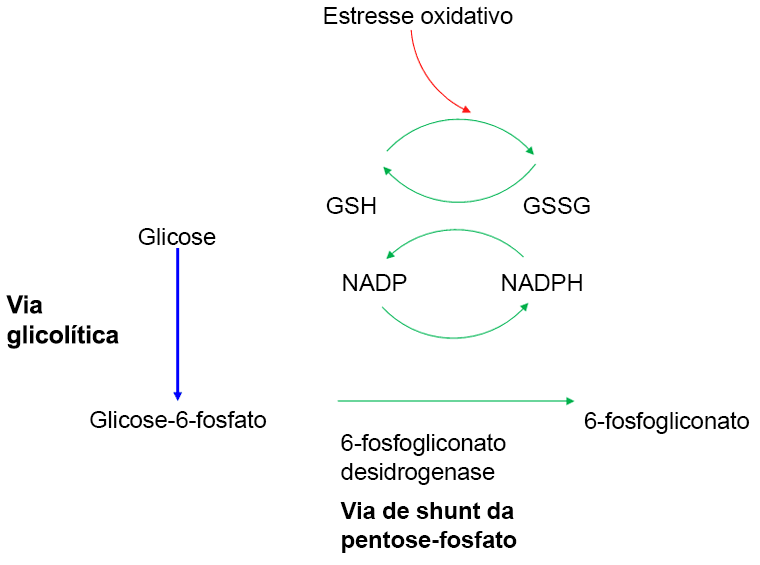
Para o eritrócito, essa é a única fonte de proteção contra o dano oxidativo. Os eritrócitos são constantemente desafiados por oxidantes, sob a forma de radicais livres gerados pela conversão de oxiemoglobina em desoxiemoglobina e pelos peróxidos gerados por granulócitos realizando a fagocitose. Os eritrócitos normais podem aumentar a geração de NADPH em resposta ao estresse oxidativo; essa capacidade é comprometida nos pacientes com deficiência de G6PD. A falha em suportar o estresse oxidativo danifica os grupos sulfidrila na hemoglobina e na membrana dos eritrócitos e provoca a hemólise. As células de outros tecidos e órgãos têm vias alternativas para a geração de NADPH e, portanto, conseguem suportar esse estresse oxidativo. Por outro lado, os eritrócitos são metabolicamente muito simples; eles não têm núcleo e mitocôndrias, não podem realizar a síntese de proteínas e metabolizam a glicose exclusivamente para a produção de adenosina trifosfato (ATP). A atividade de todas as enzimas dos eritrócitos, incluindo a G6PD, é maior nos eritrócitos jovens (reticulócitos) e declina progressivamente à medida que as células envelhecem.
Após um episódio hemolítico, a taxa de produção de eritrócitos é acelerada e a proporção elevada de eritrócitos jovens com níveis mais altos de G6PD limita a lise adicional dos eritrócitos. Portanto, os episódios hemolíticos são geralmente autolimitados em pessoas com variantes (classe III) moderadamente deficientes (por exemplo, G6PD A-), mas podem resultar em anemia mais grave e progressiva em pacientes com variantes (classe II) gravemente deficientes.
A falta completa da enzima é incompatível com a vida. Raramente, os pacientes do sexo masculino com as variantes de classe I têm atividade profundamente baixa da G6PD, de forma que os eritrócitos são constantemente submetidos à lise (anemia hemolítica não esferocítica crônica). Quase todas as pessoas com deficiência, no entanto, têm uma das variantes enzimáticas polimórficas (por exemplo, G6PD mediterrânea, classe II, ou G6PD A-, classe III) que conferem atividade residual suficiente para manter a pessoa em um estado assintomático na ausência de estresse. No entanto, quando provocadas pelo estresse oxidativo (por exemplo, ingestão de certos medicamentos, exposição a certos produtos químicos ou glicosídeos nas favas e infecção), ocorre a lise dos eritrócitos.
Sabe-se que alguns medicamentos representam um desafio oxidativo aos eritrócitos e induzem a anemia hemolítica em pacientes com deficiência de G6PD. Para alguns medicamentos (por exemplo, dapsona, primaquina, sulfonamidas), a associação é muito clara e consistente, e a hemólise será observada em quase todos os pacientes, independentemente da atividade enzimática residual.[2][11][12][13] Para outros (por exemplo, aspirina), a associação clínica é menos consistente e reflete uma interação de fatores hereditários (por exemplo, atividade enzimática residual, farmacocinética) e adquiridos (por exemplo, dose, absorção e metabolismo do medicamento, infecção coexistente).[2][11] Destacou-se a importância da hemólise induzida por primaquina na América Latina.[14] Uma revisão enfatizou que muitos compostos podem ter sido erroneamente citados como causadores de hemólise porque foram administrados em indivíduos que estavam sofrendo de hemólise relacionada a infecção.[15] Essa revisão destacou o potencial hemolítico de apenas 7 medicamentos atualmente utilizados: dapsona, azul de metileno, nitrofurantoína, fenazopiridina, primaquina, rasburicase e azul de toluidina.[Figure caption and citation for the preceding image starts]: Medicamentos e agentes conhecidos por causarem a hemólise em pessoas com deficiência de glicose-6-fosfato desidrogenase (G6PD)Criado pelo Professor Atul B. Mehta [Citation ends].
Durante o período neonatal, a hiperbilirrubinemia (icterícia fisiológica) é frequentemente exagerada em pacientes com deficiência de G6PD por causa da maior renovação de heme decorrente da lise dos eritrócitos e do comprometimento na capacidade de conjugar e depurar a bilirrubina. Esse quadro clínico é mais comum nas variantes mais graves de G6PD, e a hiperbilirrubinemia é mais grave quando a pessoa também tem uma deficiência hereditária das enzimas necessárias para conjugar a bilirrubina.[16][17][18][19][20]
Acredita-se que as anormalidades no metabolismo da bilirrubina em pacientes com deficiência de G6PD contribuam para o aumento da prevalência de colelitíase nesses pacientes.[21][22][23]
Classificação
Grupo de trabalho da Organização Mundial da Saúde (OMS) para a deficiência de glicose-6-fosfato desidrogenase (G6PD)[1]
A atividade deficiente da enzima geralmente resulta de mutações estruturais que reduzem a estabilidade da enzima. Mais de 400 enzimas variantes foram caracterizadas de acordo com as propriedades físico-químicas e bioquímicas. Essas variantes foram classificadas em 5 grupos com base na atividade enzimática e nas manifestações clínicas:
Classe I: gravemente deficiente, associada a anemia hemolítica não esferocítica crônica
Classe II: gravemente deficiente (1%-10% de atividade residual), associada a anemia hemolítica aguda
Classe III: moderadamente deficiente (10%-60% de atividade residual)
Classe IV: atividade normal (60%-150%)
Classe V: aumento da atividade (>150%).
O uso deste conteúdo está sujeito ao nosso aviso legal