Abordaje
La LMA es una enfermedad muy heterogénea. El diagnóstico de la LMA requiere un enfoque multifacético que incluya la historia clínica, la evaluación clínica y la evaluación patológica (incluida la médula ósea y/o la sangre periférica).
Dado que la LMA y la leucemia linfoblástica aguda (LLA) suelen ser clínicamente indistinguibles, es esencial confirmar el origen mieloide de las células leucémicas mediante inmunofenotipado. Esto puede realizarse en la sangre periférica antes de la confirmación a partir de la médula ósea.
Antecedentes
En todos los pacientes, una historia clínica completa (incluidos los antecedentes familiares, si se conocen) es importante para el diagnóstico.
El riesgo de LMA aumenta en determinados grupos de pacientes, como los que tienen antecedentes de enfermedad hematológica; tratamiento previo con quimioterapia; trastornos genéticos (p. ej., trastornos hereditarios de fragilidad cromosómica; síndromes de insuficiencia de la médula ósea; trisomías cromosómicas [p. ej., síndrome de Down]); edad superior a 65 años; fumadores; y exposición previa a radiación o benceno.
Las características clínicas de la anamnesis reciente que sugieren el diagnóstico de LMA están relacionados con la citopenia, e incluyen el aumento de la fatiga, los mareos, las palpitaciones, las fiebres, los sangrados de las mucosas (de las encías o de la nariz o la menorragia en las mujeres), el agrandamiento gingival, la erupción petequial y el dolor óseo. Puede haber antecedentes de erupción cutánea o masas (p. ej., cloromas cutáneos). Pueden presentarse síntomas pulmonares (p. ej., disnea) y gastrointestinales (p. ej., dolor abdominal intenso) debidos a infiltración leucémica o infección.
Un recuento de leucocitos elevado >100,000/microlitro (hiperleucocitosis) se produce en aproximadamente el 5% al 20% de los pacientes con LMA, predisponiéndoles a complicaciones como el síndrome de lisis tumoral, la afectación del sistema nervioso central (SNC) y la leucostasis (hiperleucocitosis sintomática; los síntomas incluyen dificultad respiratoria y alteración del estado mental).[3][4] Se trata de urgencias médicas que requieren tratamiento inmediato.
Exploración física
Los hallazgos pueden incluir palidez, equimosis y petequias. Pueden ser evidentes rasgos de infiltración leucémica extramedular (p. ej., hepatoesplenomegalia, linfadenopatía, masas cutáneas y testiculares). Los sitios de la infección, como absceso dental, infecciones nasofaríngeas, signos en el tórax o infecciones perianales, pueden ser aparentes. Dentro de la piel, puede haber una infiltración cutánea de leucemia, y la presencia de úlceras cutáneas (p. ej., síndrome de Sweet o piodermia gangrenosa) puede indicar neoplasia maligna subyacente. De forma infrecuente, puede presentarse abdomen agudo.
Evaluación inicial
Todos los pacientes con sospecha de LMA deben someterse a las siguientes pruebas iniciales:[24][51]
hemograma completo con diferencial
Frotis de sangre periférica
Panel metabólico completo (electrolitos séricos, perfiles renales y hepáticos, ácido úrico sérico y deshidrogenasa láctica)
Panel de coagulación (tiempo de protrombina [TP], tiempo de tromboplastina parcial activada [TTPa], fibrinógeno y dímeros D).
La mayoría de los pacientes con LMA (incluidos los que padecen leucemia promielocítica aguda [LMA], un subtipo de LMA) presentan anemia, neutropenia y/o trombocitopenia, pero el hemograma puede variar mucho. El recuento de leucocitos puede ser elevado, superando los 100,000/microlitro en aproximadamente el 5% al 20% de los pacientes con LMA.[3][4] A pesar del aumento en los leucocitos, muchos pacientes presentan una neutropenia grave (<500 granulocitos/microlitro), lo cual implica un riesgo alto de sufrir infecciones graves.
En la LMA, la placa de sangre puede mostrar blastos mieloides caracterizados por bastones de Auer o cuerpos Phi. [Figure caption and citation for the preceding image starts]: Frotis de sangre periférica de un paciente con leucemia mieloide aguda con maduración que muestra blastos mieloides con un bastón de AuerDe la colección de los doctores K. Raj y P. Mehta; utilizado con el consentimiento del paciente [Citation ends]. En la LPA, la película de sangre mostrará por lo general promielocitos hipergranulares con núcleos bilobulados y haces de bastones de Auer (así como blastos mieloides). [Figure caption and citation for the preceding image starts]: Extensión de sangre periférica de un paciente con leucemia promielocítica aguda que muestra promielocitos hipergranulares, algunos con paquetes de bastones de AuerDe la colección de los doctores K. Raj y P. Mehta; utilizado con el consentimiento del paciente [Citation ends].
En la LPA, la película de sangre mostrará por lo general promielocitos hipergranulares con núcleos bilobulados y haces de bastones de Auer (así como blastos mieloides). [Figure caption and citation for the preceding image starts]: Extensión de sangre periférica de un paciente con leucemia promielocítica aguda que muestra promielocitos hipergranulares, algunos con paquetes de bastones de AuerDe la colección de los doctores K. Raj y P. Mehta; utilizado con el consentimiento del paciente [Citation ends]. [Figure caption and citation for the preceding image starts]: Extensión de sangre periférica de un paciente con leucemia promielocítica aguda que muestra promielocitos hipergranulares con núcleos de dos lóbulos y paquetes de bastones de AuerDe la colección de los doctores K. Raj y P. Mehta; utilizado con el consentimiento del paciente [Citation ends].
[Figure caption and citation for the preceding image starts]: Extensión de sangre periférica de un paciente con leucemia promielocítica aguda que muestra promielocitos hipergranulares con núcleos de dos lóbulos y paquetes de bastones de AuerDe la colección de los doctores K. Raj y P. Mehta; utilizado con el consentimiento del paciente [Citation ends].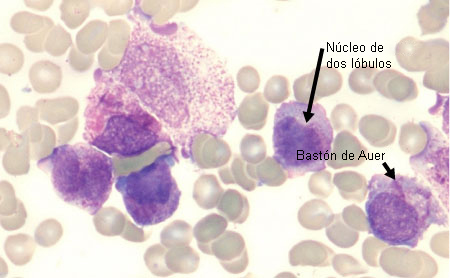 Una variante de la LPA se caracteriza por promielocitos hipogranulares (ausencia de bastones de Auer), pero es menos frecuente.
Una variante de la LPA se caracteriza por promielocitos hipogranulares (ausencia de bastones de Auer), pero es menos frecuente.
Los niveles séricos de calcio, potasio, fósforo, ácido úrico y deshidrogenasa láctica pueden estar aumentados. El grado de aumento del ácido úrico refleja la extensión de la masa tumoral. La hipercalcemia puede deberse a una infiltración ósea o a la liberación ectópica de una sustancia similar a la hormona paratiroidea.
Puede producirse hiperuricemia, hiperfosfatemia, hipocalcemia e hiperpotasemia debido al síndrome de lisis tumoral (una emergencia oncológica que requiere derivación urgente).[52] Puede producirse disfunción renal si se desarrolla un síndrome de lisis tumoral. Véase Síndrome de lisis tumoral.
Las pruebas de coagulación TP y TTPa pueden estar ligeramente prolongadas con fibrinógeno y dímero D normales. Si estas pruebas son anormales (TP y TTPa prolongados, fibrinógeno disminuido y/o dímero D elevado), debe sospecharse una coagulación intravascular diseminada (CID) y se justifica una derivación urgente para iniciar el tratamiento. Consulte el sistema de puntuación de la International Society on Thrombosis and Haemostasis (ISTH) para la coagulación intravascular diseminada (CID).[53] La CID es más frecuente en la LPA.
Debe examinarse el líquido cefalorraquídeo en los casos en los que se sospeche una afectación del SNC (es decir, recuento elevado de leucocitos, linaje monocítico o signos y síntomas neurológicos en el momento de la presentación).
Evaluación de la médula ósea
Las pruebas diagnósticas de rutina incluyen análisis de aspirado de médula ósea y biopsia por trépano.
Los frotis de médula se examinan morfológicamente. El inmunofenotipado (mediante citometría de flujo en aspirado de médula ósea) identifica los marcadores de superficie celular y citoplasmáticos de las células mieloides (p. ej., CD34 o CD33) y establece el linaje.[24] Si el aspirado de médula ósea no está disponible o es de mala calidad, puede utilizarse la inmunohistoquímica (utilizando una muestra de biopsia central) en lugar de la citometría de flujo para el inmunofenotipado. Si las muestras de médula ósea son inadecuadas o imposibles de obtener, puede utilizarse sangre periférica para la evaluación patológica siempre que haya un número suficiente de blastos circulantes.
Pruebas genéticas y moleculares
El análisis citogenético (cariotipo e hibridación fluorescente in situ [FISH]) y las pruebas genéticas moleculares informan del pronóstico y, potencialmente, del tratamiento preferido.[24][51]
En la LMA, deben investigarse las siguientes anomalías cromosómicas y mutaciones genéticas debido a su asociación con pronósticos y objetivos de tratamiento específicos: RUNX1::RUNX1T1; CBFB::MYH11; MLLT3::KMT2A (u otros reordenamientos de KMT2A); DEK::NUP214; BCR::ABL1; KAT6A:: CREBBP; NPM1; FLT3; IDH1; IDH2; CEBPA (dominio básico de cremallera de leucina [bZIP]); -5 o del(5q); -7; -17/abn(17p); GATA2; MECOM(EVI1); ASXL1; BCOR; EZH2; RUNX1; SF3B1; SRSF2; STAG2; U2AF1; ZRSR2; y TP53.[24]
Para el análisis mutacional se recomiendan los paneles de secuenciación de nueva generación y los paneles de genes multiplex.[24]
La LPA se caracteriza por la fusión LPA::RARA causada por la translocación t(15;17).[24][54]
Diagnóstico definitivo y clasificación
La LMA puede diagnosticarse y clasificarse según la última clasificación de la Organización Mundial de la Salud (OMS) (5ª edición, 2022) o la Clasificación Internacional de Consenso (ICC).[1][2] En ambas clasificaciones, los resultados de la evaluación morfológica, la evaluación de la médula ósea, las pruebas genéticas y los factores predisponentes (p. ej., tratamiento previo; neoplasias mieloides precedentes; trastornos genéticos de línea germinal) son necesarios para realizar un diagnóstico definitivo de LMA. Sin embargo, las clasificaciones difieren en la forma de clasificar subtipos específicos de LMA y en los requisitos de umbral de recuento de blastos para determinados subtipos de LMA.
La 5ª edición de la Clasificación de tumores hematolinfoides de la OMS ya no exige un umbral de recuento de blastos ≥20% para el diagnóstico de LMA con anomalías genéticas definitorias (excepto para la fusión BCR::ABL1 y la mutación CEBPA).[1] En la clasificación ICC, sin embargo, se requiere un recuento de blastos de ≥10% para diagnosticar LMA con anomalías genéticas definitorias (excepto para la fusión BCR::ABL1, en la que se requiere un recuento de blastos de ≥20%).[2]
Véase la sección Clasificación para más información.
Pruebas adicionales
La tipificación del antígeno leucocitario humano debe realizarse en todos los pacientes que se consideren para un trasplante alogénico de células madre.
La evaluación de la función cardiaca puede realizarse antes del inicio de la quimioterapia intensiva, con un ecocardiograma o una gammagrafía de adquisición múltiple. Puede realizarse una radiografía de tórax para identificar neumonía, masas mediastínicas, infiltrados pulmonares o cardiomegalia.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad