Etiología
Las alteraciones genéticas subyacen a la mayoría de los cánceres de tiroides.
En los cánceres de tiroides diferenciados (papilar, folicular), las mutaciones BRAF (fibrosarcoma de aceleración rápida homólogo B) y RAS (sarcoma de rata) o los reordenamientos RET/PTC (reordenamiento durante la transfección/cáncer de tiroides papilar) son las alteraciones genéticas más frecuentes.[26]
Los carcinomas papilares de tiroides suelen caracterizarse por mutaciones BRAF (p. ej., la sustitución BRAF V600E) y por reordenamientos RET/PTC que activan la señalización efectora a través de una proteína cinasa activada por mitógenos (MAPK).[27][28]
Los carcinomas foliculares de tiroides están asociados a mutaciones del gen RAS o presentan una translocación cromosómica que fusiona el gen de la caja apareada 8 con el gen PPAR (receptor activado por proliferadores de peroxisomas) gamma, el oncogén de fusión PAX8::PPARG.[27]
El carcinoma anaplásico de tiroides (CAT) presenta un escenario molecular heterogéneo que incluye mutaciones de TP53 (proteína tumoral P53), TERT (transcriptasa inversa de la telomerasa), BRAF y RAS.[28] Otras mutaciones en el CAT incluyen: alteraciones genéticas en la vía de la fosfatidilinositol 3-cinasa (PI3K) como las mutaciones en PTEN (homóloga de la fosfatasa y la tensina) y PI3KCA; genes implicados en la regulación epigenética como el complejo remodelador de la cromatina SWI/SNF (siglas en inglés de SWItch/Sucrose Non-Fermentable) y las histonas metiltransferasas; genes implicados en la regulación del ciclo celular (CDKN2A, CDKN2B y CCNE1); y genes de la regulación inmunitaria tumoral (PDL1, PDL2 y JAK2).[27][13][28]
El carcinoma medular tiroideo (CMT) es esporádico o hereditario (asociado con el síndrome de neoplasia endocrina múltiple [NEM] o como una forma familiar aislada).[4] Casi todos los casos de CMT hereditario, y aproximadamente el 50% de los casos de CMT esporádico, se asocian con mutaciones en el protooncogén REordenado durante la transfección (RET), que codifica un receptor de tirosina cinasa expresado en los tejidos derivados de la cresta neural.[4][29] El CMT esporádico se puede relacionar con mutaciones en RAS hasta en el 23% de los casos.[28]
El linfoma tiroideo primario está asociado a la estimulación crónica de antígenos, como se observa en la tiroiditis linfocítica crónica (enfermedad de Hashimoto) y se postula que es el resultado del desarrollo de tejido linfoide intratiroideo.[6] El subtipo de linfoma más frecuente es el linfoma difuso de células B grandes (más del 50% de los casos), seguido del linfoma del tejido linfoide asociado a las mucosas (entre el 10% y el 23%).[5] Véase los apartados Linfoma no Hodgkin y Linfoma MALT.
Fisiopatología
Los cánceres de tiroides derivan de las células epiteliales foliculares tiroideas o de las células C parafoliculares (carcinoma medular tiroideo).[14]
Las células foliculares son heterogénea, y los folículos tiroideos tienen una capacidad de crecimiento y una sensibilidad a la hormona estimulante del tiroides variables; si se producen mutaciones somáticas, los tirocitos adquieren un potencial de crecimiento diferente.[30] Los cánceres de tiroides de origen folicular pueden ser diferenciados (papilares, foliculares, oncocíticos), poco diferenciados o indiferenciados (anaplásicos).[7][8] Las células oncocíticas (anteriormente conocidas como células de Hürthle) son células foliculares tiroideas con aspecto oncocítico, caracterizadas por núcleos hipercromáticos con nucleolos prominentes y citoplasma eosinófilo granular.[31]
Las mutaciones que proporcionan una ventaja selectiva de crecimiento, favoreciendo así el desarrollo del cáncer, se identifican en más del 90% de los cánceres de tiroides.[28] La mayoría de los cánceres de tiroides albergan mutaciones en las vías de la proteína cinasa activada por mitógenos (MAPK) y de la fosfatidilinositol-3 cinasa/proteína cinasa B (PI3K/Akt).[14][28] Entre las mutaciones activadoras comunes de la vía MAPK se incluyen los reordenamientos RET/PTC y NTRK (receptor de tirosina cinasa neurotrófico), y las mutaciones RAS y BRAF.[28]
El comportamiento fisiológico depende del tipo de tumor
Se cree que el cáncer de tiroides refleja un continuo que va desde el bien diferenciado hacia el anaplásico y que se caracteriza por fenómenos genéticos tempranos y tardíos.[32]
El carcinoma de tiroides poco diferenciado (PDTC, por sus siglas en inglés) es una neoplasia folicular que muestra evidencia limitada de diferenciación de células foliculares. Se sitúa morfológicamente y en cuanto a su comportamiento entre los carcinomas diferenciados (papilar, folicular, oncocítico) y los carcinomas anaplásicos de tiroides. Los cánceres de tiroides diferenciados o PDTC que pierden su capacidad de absorber y concentrar el yodo radioactivo (y, en algunos casos, de producir tiroglobulina) durante el avance del tumor se denominan comúnmente cánceres de tiroides desdiferenciados.[33] Hasta dos tercios de los pacientes con cáncer diferenciado de tiroides presentan una desdiferenciación tumoral.[34]
El carcinoma papilar tiende a extenderse a los ganglios linfáticos locales, mientras que el carcinoma folicular y el carcinoma oncocítico se extienden más a menudo por vía hematógena. El carcinoma anaplásico de tiroides es agresivo con una alta propensión a la invasión local y a la diseminación metastásica. La diseminación ganglionar es frecuente en los linfomas tiroideos primarios.[Figure caption and citation for the preceding image starts]: Radiografía de cráneo que muestra metástasis extensa de un carcinoma folicular de tiroidesWani AM, Hussain WM, Fatani MI, et al. Skull metastases from thyroid carcinoma. BMJ Case Reports. 2009; doi:10.1136/bcr.02.2009.1578 [Citation ends].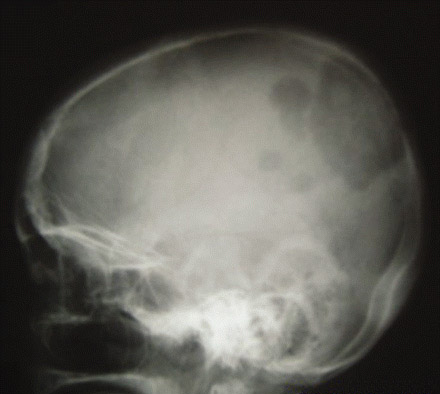
El carcinoma medular de tiroides (CMT) es esporádico (75%) o hereditario (25%).[28] El CMT hereditario puede darse como un componente de la neoplasia endocrina múltiple (NEM) tipo 2A (NEM2A) o NEM2B, o puede darse en una forma familiar aislada.[4] Casi todos los pacientes con NEM2A, NEM2B o CMT familiar aislado tienen mutaciones de la línea germinal en el protooncogén RET (que codifica un receptor de tirosina cinasa expresado en tejidos derivados de la cresta neural), y cerca del 50% de los CMT esporádicos tienen mutaciones somáticas en RET.[4] Las mutaciones de RAS están presentes en hasta el 23% de los casos esporádicos de CMT.[28] En los pacientes con CMT esporádico, la presencia de mutaciones RET se asocia a un peor pronóstico (mayor estadificación tumoral, una categoría T más alta y la presencia de metástasis en los ganglios linfáticos y a distancia); aquellos con mutaciones RAS presentaban un fenotipo menos agresivo y un mejor pronóstico.[35]
Clasificación
Tipos de cáncer de tiroides
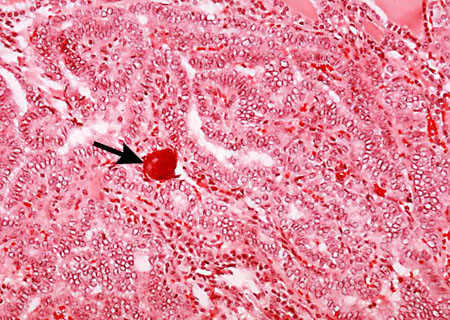
Papilar: es el tipo más común y representa el 80% de los cánceres de tiroides.[1] Generalmente, está bien diferenciado, tiende a ser multicéntrico y a afectar a los ganglios linfáticos.[Figure caption and citation for the preceding image starts]: Histopatología de un carcinoma papilar de tiroides: se puede ver un cuerpo de psamoma (flecha)CDC Image Library/Dr Edwin P. Ewing, Jr [Citation ends].
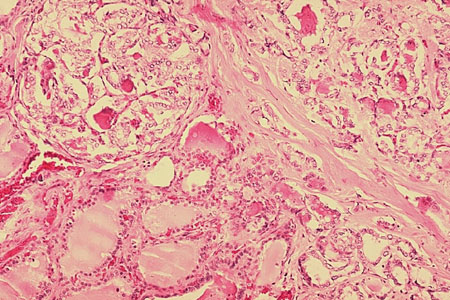
Folicular: representa aproximadamente el 10% de los cánceres de tiroides.[1] Los carcinomas foliculares de tiroides son tumores foliculares malignos con invasión capsular y/o vascular. Se propagan por invasión directa hematógena en lugar de hacerlo a través de los ganglios linfáticos. Las formas incipientes son indolentes, mientras que las formas de invasión generalizada son agresivas.[Figure caption and citation for the preceding image starts]: Histopatología de carcinoma folicular de tiroidesCDC Image Library/Dr Edwin P. Ewing, Jr [Citation ends].
Oncocítico (anteriormente conocido como célula de Hürthle): representa entre el 3% y el 4% de los cánceres de tiroides.[2] Las células oncocíticas tienen un aspecto citológico clásico de células grandes con abundantes citoplasmas granulares eosinófilos y grandes núcleos hipercromáticos con nucléolos prominentes. Los carcinomas oncocíticos de tiroides son tumores malignos con invasión capsular y/o vascular, metástasis ganglionares o metástasis a distancia y se consideran más agresivos que los carcinomas foliculares de tiroides no oncocíticos.[2]
[Figure caption and citation for the preceding image starts]: Células oncocíticas (anteriormente conocidas como células de Hürthle) con abundante citoplasma granular eosinófilo y nucleolos de color "rosa cereza"Sandoval MAS et al. Case Reports 2011;2011:bcr1120103536; usado con autorización [Citation ends].
 [Figure caption and citation for the preceding image starts]: Carcinoma oncocítico (anteriormente conocido como carcinoma de células de Hürthle): presencia de células tumorales dentro de una vena, indicativo de invasión vascularSandoval MAS et al. Case Reports 2011;2011:bcr1120103536; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Carcinoma oncocítico (anteriormente conocido como carcinoma de células de Hürthle): presencia de células tumorales dentro de una vena, indicativo de invasión vascularSandoval MAS et al. Case Reports 2011;2011:bcr1120103536; usado con autorización [Citation ends].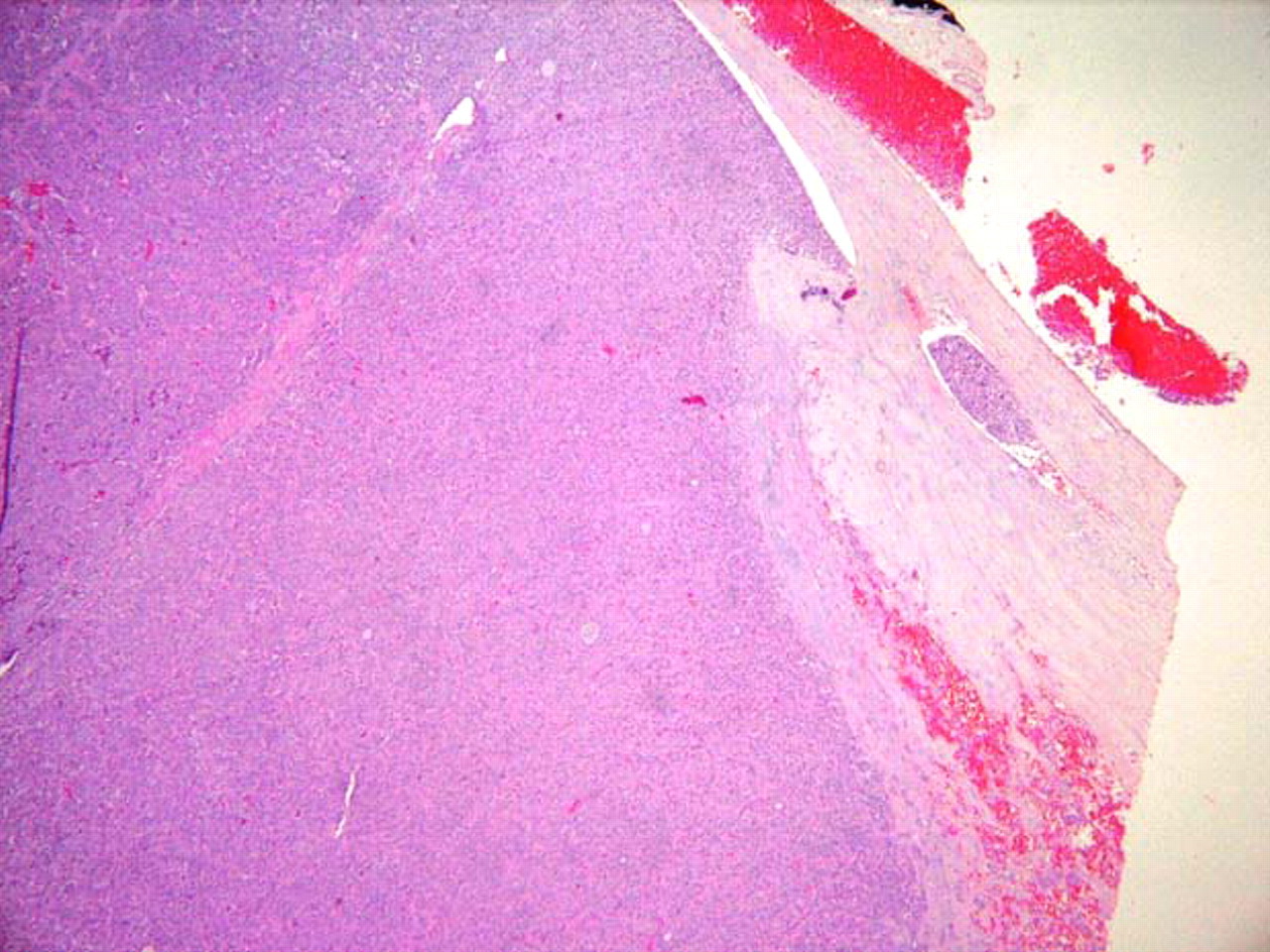
Anaplásico: neoplasia indiferenciada con invasión vascular. Generalmente presenta intrusión local dentro del nervio laríngeo recurrente y la tráquea, el músculo o el esófago.[3]
Medular: se origina en las células C parafoliculares del tiroides y representa aproximadamente del 1% al 3% de los cánceres de tiroides.[4] Se da de forma esporádica o puede ser hereditario. Una minoría (aproximadamente una cuarta parte) de los casos son hereditarios; por ejemplo, parte de los síndromes de neoplasia endocrina múltiple (NEM). Tiende a ser multicéntrico y a propagarse de manera temprana a los ganglios linfáticos.[Figure caption and citation for the preceding image starts]: Cáncer medular de tiroides: tinción H&E que muestra nidos de células tumoralesMohan V et al. BMJ Case Reports CP 2019;12:e230446; usado con autorización [Citation ends].
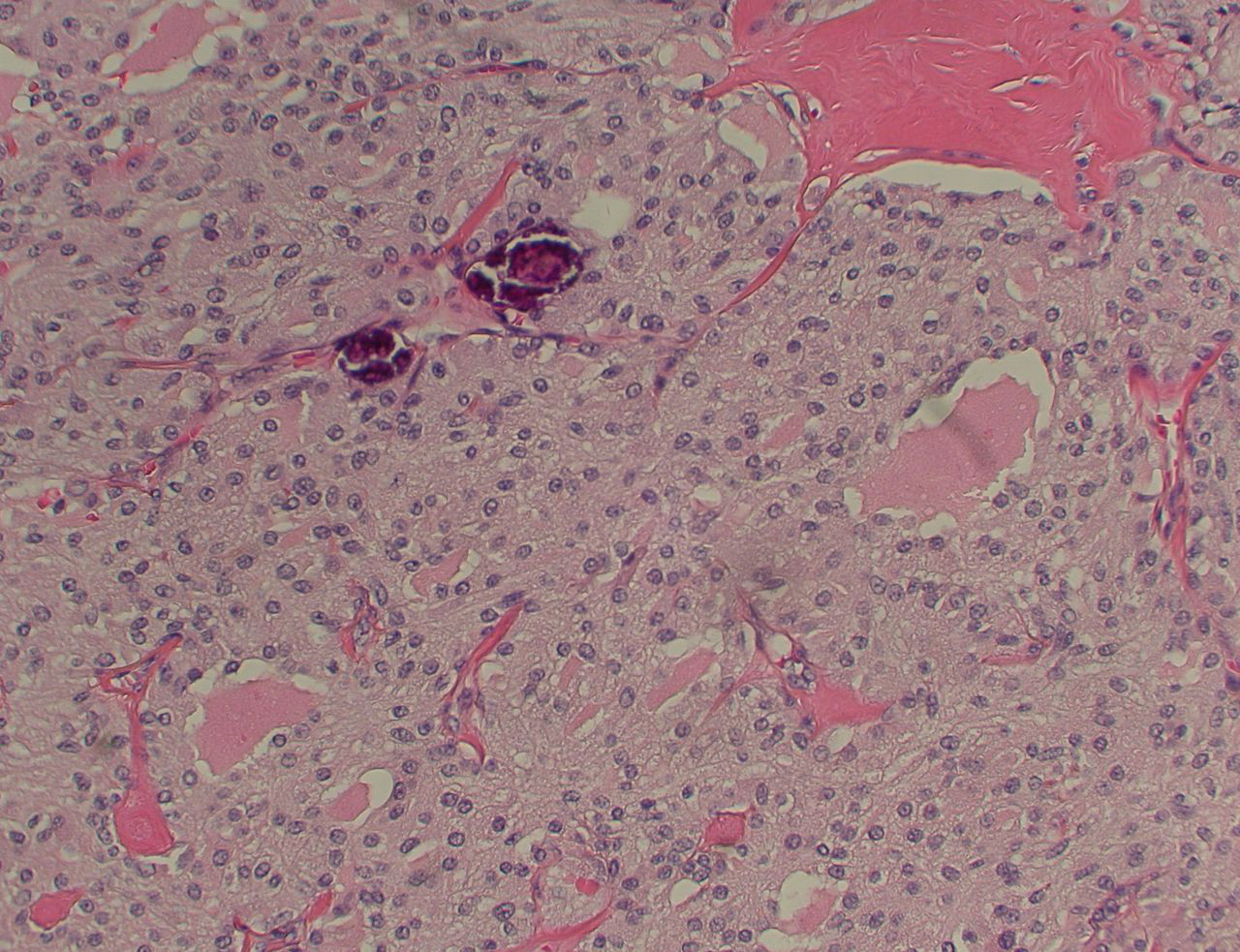 [Figure caption and citation for the preceding image starts]: Carcinoma medular tiroideo: tinción de calcitoninaMohan V et al. BMJ Case Reports CP 2019;12:e230446; usado con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Carcinoma medular tiroideo: tinción de calcitoninaMohan V et al. BMJ Case Reports CP 2019;12:e230446; usado con autorización [Citation ends].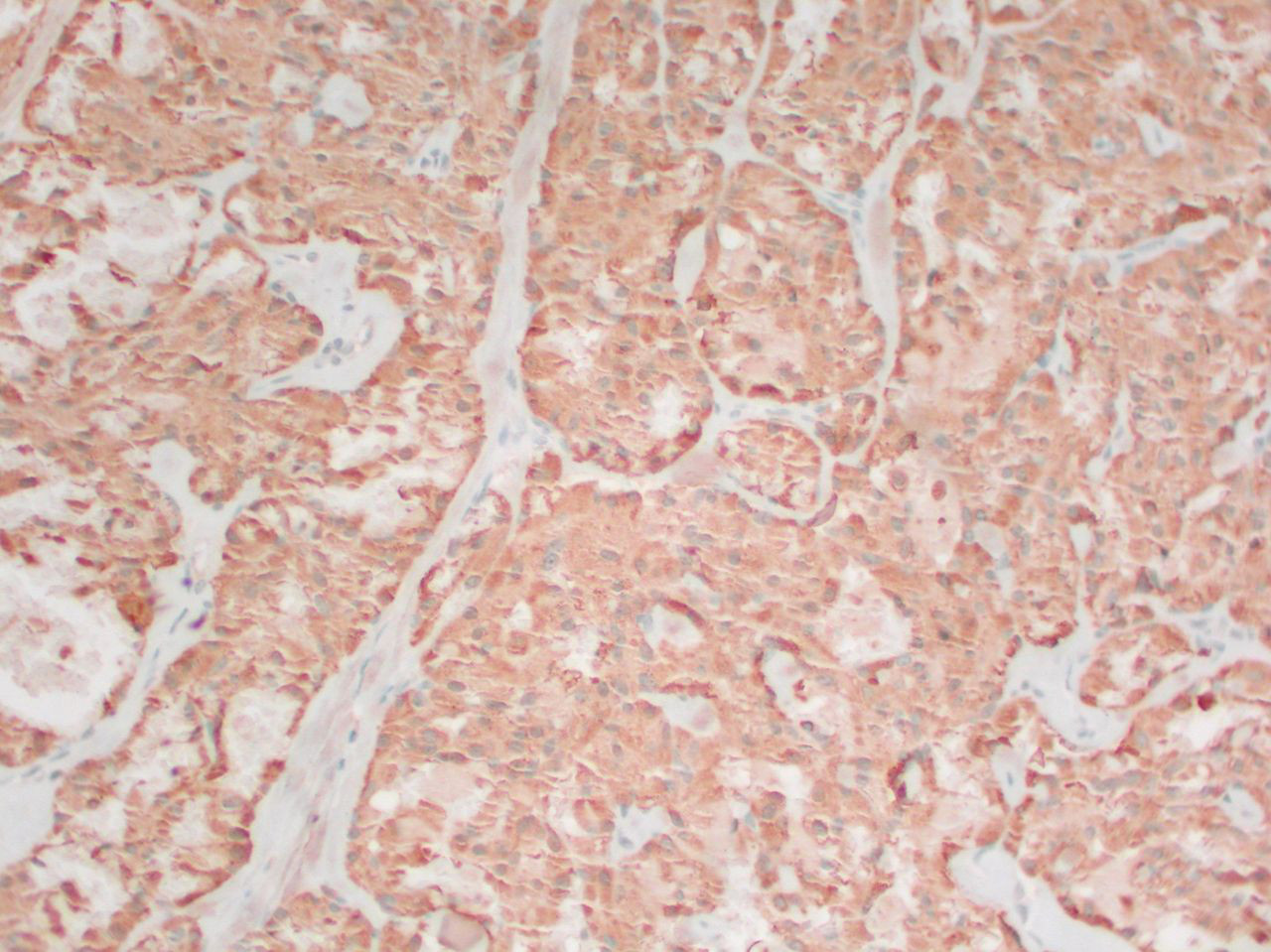
Linfoma: por lo general es un linfoma no Hodgkin de linfocitos B.[5] Generalmente surge en el contexto de una tiroiditis de Hashimoto preexistente.[6] Véase los apartados Linfoma no Hodgkin y Linfoma MALT.
Organización Mundial de la Salud (OMS) 2022: tumores de tiroides[7]
Neoplasias derivadas de células foliculares
Tumores benignos
Enfermedad folicular nodular
Adenoma folicular
Adenoma folicular con arquitectura papilar
Adenoma oncocítico
Neoplasias de bajo riesgo
Neoplasia folicular no invasiva con características nucleares de tipo papilar
Tumores de potencial maligno incierto
Tumor folicular de potencial maligno incierto
Tumor bien diferenciado de potencial maligno incierto
Tumor trabecular hialinizante
Neoplasias malignas
Carcinoma folicular
Carcinoma papilar de variante folicular encapsulado invasivo
Carcinoma papilar
Carcinoma oncocítico
Carcinomas de origen folicular de alto grado
Carcinoma poco diferenciado
Carcinoma diferenciado de alto grado
Carcinoma anaplásico derivado de células foliculares
Carcinoma de tiroides derivado de células C
Carcinoma medular de tiroides
El sistema Bethesda 2023 para la notificación de la citopatología tiroidea[8]
Un sistema de notificación estandarizado con seis categorías diagnósticas para las muestras de aspiración con aguja fina (AAF) de tiroides. Cada categoría presenta un riesgo implícito de cáncer que recomienda el siguiente paso de manejo clínica.
Categorías diagnósticas basadas en la citología AAF:
No diagnóstico
Solo líquido del quiste
Muestra virtualmente acelular
Otros (oscurecimiento de sangre, artefacto de coagulación, artefacto de secado, etc.)
Benigna
Compatible con enfermedad folicular nodular (incluye nódulo adenomatoide, nódulo coloideo, etc.)
Compatible con tiroiditis linfocítica crónica (Hashimoto) en el contexto clínico adecuado
Compatible con tiroiditis granulomatosa (subaguda)
Otros
Atipia de significado indeterminado (AUS)
Especifique si es AUS-atipia nuclear o AUS-otra
Neoplasia folicular
Especifique si es de tipo oncocítico (anteriormente célula de Hürthle)
Sospecha de neoplasia maligna
Sospecha de carcinoma papilar de tiroides
Sospecha de carcinoma medular de tiroides
Sospecha de carcinoma metastásico
Sospecha de linfoma
Otros
Derrames malignos
Carcinoma papilar de tiroides
Carcinoma folicular de alto grado
Carcinoma medular de tiroides
Carcinoma indiferenciado (anaplásico)
Carcinoma de células escamosas
Carcinoma con características mixtas (especificar)
Neoplasia maligna
Linfoma no Hodgkin
Otros
Estadificación TNM del cáncer de tiroides[9][10]
La clasificación de estadificación TNM del American Joint Committee on Cancer (AJCC) y la Union for International Cancer Control (UICC) para el carcinoma de tiroides diferenciado, anaplásico y medular se basa en los siguientes factores anatómicos:
Tamaño y extensión del tumor primario (T)
Afectación de los ganglios linfáticos regionales (N)
Presencia o ausencia de metástasis a distancia (M)
La estadificación del carcinoma diferenciado de tiroides se clasifica además por edad, en menores de 55 años y mayores de 55 años.
American College of Radiology Thyroid Imaging, Reporting, and Data System (ACR TI-RADS)[11]
Un sistema de estratificación del riesgo para los nódulos tiroideos basado en las características de la ecografía (composición, ecogenicidad, forma, margen, focos ecogénicos, tamaño del nódulo) para orientar las decisiones sobre la aspiración con aguja fina (AAF) o el seguimiento.
Composición (elija 1)
Quístico o casi completamente quístico: 0 puntos
Espongiforme: 0 puntos
Mezcla quística y sólida: 1 punto
Sólido o casi completamente sólido: 2 puntos
Ecogenicidad (elija 1)
Anecoico: 0 puntos
Hiperecoico o isoecoico: 1 punto
Hipoecoico: 2
Muy hipoecoico: 3 puntos
Forma (elija 1)
Más ancho que alto: 0 puntos
Más alto que ancho: 3 puntos
Margen (elija 1)
Suave: 0 puntos
Mal definido: 0 puntos
Lobulado o irregular: 2 puntos
Extensión extratiroidea: 3 puntos
Focos ecogénicos (elija todos los que correspondan)
Nada o grandes artefactos de cola de cometa: 0 puntos
Macrocalcificaciones: 1 punto
Calcificaciones periféricas (borde): 2 puntos
Focos ecogénicos puntiformes: 3 puntos
El número total de puntos de todas las características (categorías) de la ecografía determina la estratificación del riesgo TI-RADS (TR1 a TR5)
TR1 (benigno): 0 puntos
TR2 (no sospechoso): 2 puntos
TR3 (levemente sospechoso): 3 puntos
TR4 (moderadamente sospechoso): 4 a 6 puntos
TR5 (altamente sospechoso): 7 puntos o más
Las recomendaciones para la AAF o el seguimiento se basan en la estratificación del riesgo de TI-RADS y el tamaño del nódulo
TR1: no realizar AAF
TR2: no realizar AAF
TR3: AAF si el nódulo mide ≥2,5 cm, o realizar seguimiento si mide ≥1,5 cm
TR4: AAF si el nódulo mide ≥1,5 cm, o realizar seguimiento si mide ≥1 cm
TR5: AAF si el nódulo mide ≥1 cm, o seguir si mide ≥0.5 cm. La AAF de nódulos TR5 con de 0.5 a 0.9 cm pueden ser adecuados para los microcarcinomas papilares en determinadas circunstancias
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad