Etiología
La exposición a corticosteroides exógenos es la causa más común del síndrome de Cushing.
La mayoría (70% a 80%) de los pacientes con síndrome de Cushing endógeno presentan adenomas hipofisarios secretores de hormona adrenocorticotropa (ACTH); sin embargo, solo el 10% de todos los adenomas hipofisarios secretan un exceso de ACTH.[8][16] Los adenomas corticotropos silenciosos son inmunopositivos para la ACTH, pero se presentan clínicamente como adenomas no funcionales. Sin embargo, los adenomas de gran tamaño se convierten con escasa frecuencia en secretores de ACTH y causan la enfermedad de Cushing.[17] Se descubrió que un nuevo gen, el USP8 (proteasa ubiquitina específica 8), está frecuentemente mutado en la enfermedad de Cushing.[18]
Alrededor del 10% de los pacientes con síndrome de Cushing endógeno presentan adenomas suprarrenales con una secreción no regulada de cortisol, pero solo el 5% de todos los adenomas suprarrenales desarrollan una secreción autónoma de cortisol.[8][14] Las mutaciones en la vía monofosfato de adenosina cíclica (cAMP)/proteína cinasa A son la causa de la mayoría de los casos de hipercortisolismo suprarrenal primario.[19] No se identificaron exposiciones específicas ni factores modificables que causen hipercortisolismo endógeno. No se conoce bien la etiología de la sobreproducción de ACTH del adenoma hipofisario y la sobreproducción de cortisol del adenoma suprarrenal.
Solo alrededor del 1% de los pacientes con síndrome de Cushing presentan un carcinoma suprarrenal. Por otro lado, la superproducción suprarrenal de cortisol se observa entre el 50% y el 60% de los carcinomas suprarrenales, lo cual genera un síndrome de Cushing independiente de la ACTH.[10]
Fisiopatología
Las manifestaciones clínicas son consecuencia de la exposición excesiva del tejido al cortisol. El nivel en el que se manifiestan los síntomas se basa en gran medida, si no totalmente, en el nivel de exceso de cortisol. Los pacientes con hipercortisolismo leve a moderado generalmente presentan un fenotipo menos prominente con intolerancia a la glucosa, dislipidemia, osteopatía metabólica y aumento de peso anómalo, pero son difíciles de diferenciar de otros pacientes con el síndrome metabólico. A medida que el hipercortisolismo aumenta, las características físicas empeoran con desarrollo de estrías, almohadillas de grasa supraclaviculares y debilidad muscular proximal. La secreción de hormona adrenocorticotropa (ACTH) ectópica de los tumores neuroendocrinos puede manifestarse como casos más graves con la presentación abrupta de síntomas y niveles muy elevados de cortisol y de ACTH. Estos pacientes también pueden tener debilidad muscular y pérdida de peso graves.
Clasificación
Etiologías del hipercortisolismo
Dependiente de la hormona adrenocorticotropa (ACTH)
Causado por enfermedades que van acompañadas de niveles altos, o inadecuadamente normales, de hormona adrenocorticotrópica (ACTH) y que estimulan la sobreproducción de cortisol suprarrenal.
Los adenomas hipofisarios secretores de ACTH (enfermedad de Cushing) y los tumores secretores de ACTH ectópica son dos formas de la enfermedad dependiente de la ACTH. Los tumores secretores de ACTH ectópica son generalmente de origen broncogénico o neuroendocrino.
La hormona liberadora de corticotropina ectópica puede considerarse dentro de esta categoría, pero es extremadamente poco común y aquí no se analiza específicamente en detalle.
Independiente de la ACTH
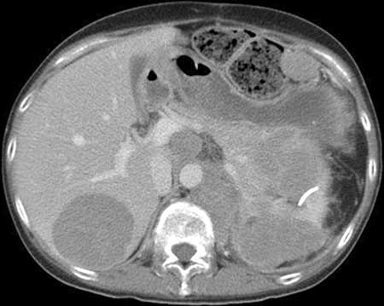
El síndrome de Cushing independiente de la ACTH lo causa la secreción excesiva de cortisol de las glándulas suprarrenales a pesar de un nivel de ACTH suprimido. En esta categoría se incluyen los adenomas suprarrenales, la hiperplasia suprarrenal bilateral y, rara vez, el carcinoma suprarrenal. El carcinoma suprarrenal es extremadamente raro y se presenta típicamente como una masa suprarrenal grande (>5 cm) y de rápido crecimiento.[Figure caption and citation for the preceding image starts]: Exploración computarizada del abdomen que muestra un tumor adrenocortical infiltrado en el páncreas y el riñón izquierdo, y con metástasis en el hígado, el bazo y los ganglios centralesDel BMJ Case Reports 2010; doi:10.1136/bcr.07.2009.2100 [Citation ends].
La hiperplasia suprarrenal macronodular bilateral ACTH independiente (AIMAH) es una forma poco común de enfermedad suprarrenal nodular bilateral. La fisiopatología de la AIMAH es la estimulación de receptores aberrantes. Se han identificado al menos 10 receptores aberrantes diferentes que causan la AIMAH. Estos incluyen el receptor del polipéptido inhibidor gástrico, los receptores beta adrenérgicos, el receptor de vasopresina, el receptor de serotonina, el receptor de angiotensina II, el receptor de hormona luteinizante y gonadotropina coriónica humana, y el receptor de leptina.[2] La estimulación de estos receptores conduce al crecimiento inadecuado de nódulos bilaterales, grandes, monoclonales y policlonales.[3][4] Clínicamente, la AIMAH se presenta con mayor frecuencia en la quinta y la sexta décadas de vida con exceso de producción de glucocorticoides y mineralocorticoides.[5] En los estudios por imágenes, se observan glándulas suprarrenales masivamente agrandadas de forma bilateral.
La enfermedad adrenocortical nodular pigmentada primaria (PPNAD), que generalmente es parte del complejo de Carney (un síndrome de neoplasia endocrina múltiple caracterizado por lentigos, mixomas cardíacos y cutáneos, y tumores endocrinos; puede ser hereditario de una manera autosómica dominante), es una afección poco frecuente, causada por numerosos nódulos suprarrenales pequeños y de funcionamiento autónomo cuyo tamaño varía desde el tamaño microscópico a 1 cm. En la exploración patológica, los nódulos presentan pigmentación oscura y la corteza suprarrenal interviniente es atrófica. Se cree que las mutaciones en la subunidad reguladora de la proteína cinasa A tipo 1 alfa (PRKAR1A) son las mutaciones causales. La PPNAD tiene una edad de distribución bimodal, y la mayoría de los casos se diagnostican en la segunda y la tercera décadas de vida.[6] Los pacientes con PPNAD, por lo general, son delgados y no presentan la obesidad central típica que se observa en otras causas del síndrome de Cushing. Osteoporosis grave, estatura baja y pérdida de masa muscular grave son las características comunes que presentan los pacientes con PPNAD.[7]
Exógenas
Los pacientes que toman corticosteroides exógenos, por algún motivo, pueden desarrollar características del síndrome de Cushing. Cuando se suspenden las dosis altas de glucocorticosteroides, los pacientes pueden desarrollar insuficiencia suprarrenal a pesar del fenotipo clínico de Cushing.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad