A abordagem diagnóstica inclui a obtenção de uma história completa dos principais sintomas diagnósticos, como uma massa indolor e unilateral na parte superior do abdome/no flanco, síndromes congênitas e anomalias urogenitais congênitas.[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
A ultrassonografia abdominal é o exame inicial de escolha para estabelecer o diagnóstico, e a tomografia computadorizada (TC) ou a ressonância nuclear magnética (RNM) abdominal e pélvica são usadas para estadiar o tumor e planejar a continuação da terapia. O diagnóstico definitivo da suspeita de tumor de Wilms é feito com base na histologia do tumor seguida por ressecção cirúrgica (nefrectomia) ou biópsia (se o tumor for irressecável). Deve-se descartar a presença de doença metastática na TC do tórax (ou radiografia torácica em áreas com recursos limitados) e TC ou RNM abdominal/pélvica.
História
História familiar de tumor de Wilms e presença de anomalias geniturinárias congênitas devem ser documentadas.[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Anomalias fenotípicas específicas que estão associadas a síndromes relacionadas ou não ao supercrescimento devem ser descartadas.[17]PDQ Pediatric Treatment Editorial Board. Wilms tumor and other childhood kidney tumors treatment (PDQ®): health professional version. 2022 Dec 23. In: PDQ cancer information summaries [internet]. Bethesda, MD: National Cancer Institute; 2002.
https://www.cancer.gov/types/kidney/hp/wilms-treatment-pdq#_1
http://www.ncbi.nlm.nih.gov/pubmed/26389282?tool=bestpractice.com
Por exemplo, hipoglicemia hiperinsulinêmica, que pode ser transitória ou persistente, ocorre em 50% das crianças com síndrome de Beckwith-Wiedemann durante o período neonatal e a primeira infância; portanto, se houver suspeita dessa síndrome, história de hipoglicemia no nascimento deverá ser observada.[35]Green DM, Breslow NE, Beckwith JB, et al. Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol. 1993;21(3):188-92.
http://www.ncbi.nlm.nih.gov/pubmed/8095320?tool=bestpractice.com
O tumor de Wilms é mais comum em crianças negras e brancas em comparação com crianças asiáticas e ocorre mais comumente nos primeiros 5 anos de vida.[1]Nakata K, Colombet M, Stiller CA, et al. Incidence of childhood renal tumours: an international population-based study. Int J Cancer. 2020 Dec 15;147(12):3313-27.
https://onlinelibrary.wiley.com/doi/10.1002/ijc.33147
http://www.ncbi.nlm.nih.gov/pubmed/32902866?tool=bestpractice.com
[2]Szychot E, Apps J, Pritchard-Jones K. Wilms' tumor: biology, diagnosis and treatment. Transl Pediatr. 2014 Jan;3(1):12-24.
https://tp.amegroups.com/article/view/3228/html
http://www.ncbi.nlm.nih.gov/pubmed/26835318?tool=bestpractice.com
O tumor se manifesta com mais frequência como uma massa indolor e unilateral na parte superior do abdome/no flanco.[2]Szychot E, Apps J, Pritchard-Jones K. Wilms' tumor: biology, diagnosis and treatment. Transl Pediatr. 2014 Jan;3(1):12-24.
https://tp.amegroups.com/article/view/3228/html
http://www.ncbi.nlm.nih.gov/pubmed/26835318?tool=bestpractice.com
Os tumores bilaterais são raros e ocorrem em cerca de 10% dos pacientes, podendo se manifestar simultaneamente em ambos os rins (síncronos) ou comprometer um rim seguido do outro (metácronos).[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
[5]Charlton J, Irtan S, Bergeron C, et al. Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med. 2017 Jul 18;19:e8.
https://www.cambridge.org/core/journals/expert-reviews-in-molecular-medicine/article/bilateral-wilms-tumour-a-review-of-clinical-and-molecular-features/B4C8FBEE8C2C189D7D739B9BD64F1AA8
http://www.ncbi.nlm.nih.gov/pubmed/28716159?tool=bestpractice.com
Outras características clínicas, como palidez, dor abdominal, febre, hematúria (visível ou não), inapetência e perda de peso podem estar presentes.[2]Szychot E, Apps J, Pritchard-Jones K. Wilms' tumor: biology, diagnosis and treatment. Transl Pediatr. 2014 Jan;3(1):12-24.
https://tp.amegroups.com/article/view/3228/html
http://www.ncbi.nlm.nih.gov/pubmed/26835318?tool=bestpractice.com
[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
[17]PDQ Pediatric Treatment Editorial Board. Wilms tumor and other childhood kidney tumors treatment (PDQ®): health professional version. 2022 Dec 23. In: PDQ cancer information summaries [internet]. Bethesda, MD: National Cancer Institute; 2002.
https://www.cancer.gov/types/kidney/hp/wilms-treatment-pdq#_1
http://www.ncbi.nlm.nih.gov/pubmed/26389282?tool=bestpractice.com
A presença de dispneia pode estar associada a anemia ou metástase pulmonar.
Muito raramente, o tumor de Wilms ocorre em localizações extrarrenais.[6]Shojaeian R, Hiradfar M, Sharifabad PS, et al. Extrarenal Wilms’ tumor: challenges in diagnosis, embryology, treatment and prognosis. In: van den Heuvel-Eibrink MM, ed. Wilms tumor [internet]. Brisbane: Codon Publications; 2016 Mar: chapter 6.
https://www.ncbi.nlm.nih.gov/books/NBK373353
http://www.ncbi.nlm.nih.gov/pubmed/27512762?tool=bestpractice.com
A manifestação (massa indolor de crescimento rápido) é geralmente semelhante à do tumor de Wilms clássico, mas os sinais e sintomas são exclusivos do local do tumor.
Exame físico
O local e a extensão da massa abdominal devem ser documentados. A massa geralmente é retroperitoneal ("balão") e não se move com a respiração.[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
É lisa, firme ao toque e insensível à palpação. O paciente pode ter distensão abdominal. A genitália deve ser examinada quanto à presença de anormalidades geniturinárias congênitas (isto é, hipospádia, genitália atípica ou criptorquidia).[16]Narod SA, Hawkins MM, Robertson CM, et al. Congenital anomalies and childhood cancer in Great Britain. Am J Hum Genet. 1997 Mar;60(3):474-85.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1712528
http://www.ncbi.nlm.nih.gov/pubmed/9042906?tool=bestpractice.com
[29]Breslow NE, Beckwith JB. Epidemiological features of Wilms' tumor: results of the National Wilms' Tumor Study. J Natl Cancer Inst. 1982 Mar;68(3):429-36.
http://www.ncbi.nlm.nih.gov/pubmed/6278194?tool=bestpractice.com
A presença de uma varicocele na posição supina pode estar associada à extensão do tumor na veia cava inferior ou na veia renal.[36]Idowu BM, Tanimola AG. Wilm's tumor presenting with scrotal varicocele in an 11-month-old boy. Indian J Radiol Imaging. 2018 Apr-Jun;28(2):247-9.
https://www.thieme-connect.com/products/ejournals/abstract/10.4103/ijri.IJRI_279_17
http://www.ncbi.nlm.nih.gov/pubmed/30050251?tool=bestpractice.com
A hipertensão está presente em aproximadamente 25% dos pacientes e é secundária à compressão da vasculatura renal ou devida à hipersecreção de renina.[4]Davidoff AM. Wilms tumor. Adv Pediatr. 2012;59(1):247-67.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3589819
http://www.ncbi.nlm.nih.gov/pubmed/22789581?tool=bestpractice.com
[37]Maas MH, Cransberg K, van Grotel M, et al. Renin-induced hypertension in Wilms tumor patients. Pediatr Blood Cancer. 2007 May;48(5):500-3.
http://www.ncbi.nlm.nih.gov/pubmed/16794999?tool=bestpractice.com
A hepatomegalia pode indicar doença metastática.[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
A extensão intracardíaca do tumor de Wilms é rara.[10]Abdullah Y, Karpelowsky J, Davidson A, et al. Management of nine cases of Wilms' tumour with intracardiac extension - a single centre experience. J Pediatr Surg. 2013 Feb;48(2):394-9.
http://www.ncbi.nlm.nih.gov/pubmed/23414872?tool=bestpractice.com
As anormalidades fenotípicas que podem ser características das síndromes associadas ao tumor de Wilms devem ser documentadas.[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Raramente, as crianças podem apresentar uma síndrome paraneoplásica que afeta o sistema nervoso central e periférico (por exemplo, fraqueza generalizada, fadiga, ptose, hipocinesia, disartria, retenção urinária, diplegia facial, oftalmoplegia e disfunção autonômica).[9]Petersen CL, Hemker BG, Jacobson RD, et al. Wilms tumor presenting with lambert-eaton myasthenic syndrome. J Pediatr Hematol Oncol. 2013 May;35(4):267-70.
http://www.ncbi.nlm.nih.gov/pubmed/23612377?tool=bestpractice.com
Investigações laboratoriais
Devem ser solicitados exames da função renal e hepática, hemograma completo, urinálise, proteínas séricas/albumina e estudos de coagulação, embora eles não sejam necessários para o diagnóstico.
Exames por imagem
Os estudos iniciais visam estabelecer a origem renal e a extensão da massa. A ultrassonografia abdominal é o exame de primeira linha recomendado para estabelecer o diagnóstico presuntivo e geralmente é adequada para esse fim.[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
[38]van der Beek JN, Artunduaga M, Schenk JP, et al. Similarities and controversies in imaging of pediatric renal tumors: a SIOP-RTSG and COG collaboration. Pediatr Blood Cancer. 2022 Nov 9;e30080.
http://www.ncbi.nlm.nih.gov/pubmed/36349564?tool=bestpractice.com
O achado típico é uma grande massa intrarrenal ecogênica, heterogênea, unilateral e principalmente sólida (embora pequenas áreas de alterações císticas possam ser observadas). Se houver suspeita de tumor de Wilms na ultrassonografia, o paciente deverá ser imediatamente encaminhado a um grande centro de câncer pediátrico onde novos exames por imagem serão planejados.
Tanto uma TC quanto uma RNM abdominal e pélvica devem ser realizadas para estadiamento locorregional, avaliando o rim contralateral quanto à presença de doença síncrona e determinando o tamanho e o número de massas ipsilaterais, presença de linfadenopatia, sinais de possível ruptura, infiltração em órgãos adjacentes, presença e extensão de trombo tumoral e presença de doença metastática em órgãos como o fígado.[38]van der Beek JN, Artunduaga M, Schenk JP, et al. Similarities and controversies in imaging of pediatric renal tumors: a SIOP-RTSG and COG collaboration. Pediatr Blood Cancer. 2022 Nov 9;e30080.
http://www.ncbi.nlm.nih.gov/pubmed/36349564?tool=bestpractice.com
O protocolo da International Society of Paediatric Oncology (SIOP), bem como o protocolo do Children's Oncology Group (COG), usam a TC de tórax para detecção de lesões pulmonares no momento do diagnóstico.[38]van der Beek JN, Artunduaga M, Schenk JP, et al. Similarities and controversies in imaging of pediatric renal tumors: a SIOP-RTSG and COG collaboration. Pediatr Blood Cancer. 2022 Nov 9;e30080.
http://www.ncbi.nlm.nih.gov/pubmed/36349564?tool=bestpractice.com
Em regiões com recursos limitados, pode-se usar a radiografia de tórax para identificar metástases pulmonares; no entanto, a radiografia simples pode não mostrar lesões pulmonares menores (normalmente <1 cm).[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
O papel da tomografia por emissão de pósitrons no estadiamento e na avaliação da resposta não está claro.[39]Provenzi M, Saettini F, Conter V, et al. Is there a role for FDG-PET for the assessment of treatment efficacy in Wilms' tumor? A case report and literature review. Pediatr Hematol Oncol. 2013 Oct;30(7):633-9.
http://www.ncbi.nlm.nih.gov/pubmed/24050763?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Tumor de Wilms: achados da RNMUHRAD.com; usado com permissão [Citation ends].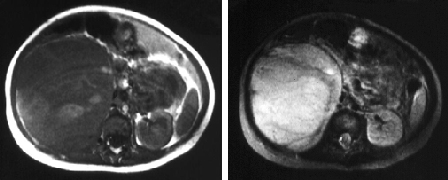
Histologia tumoral
O diagnóstico definitivo se baseia na histologia do tumor seguida por ressecção cirúrgica (nefrectomia) ou biópsia (se o tumor for irressecável).[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
No protocolo do COG, é necessária uma biópsia aberta com, no mínimo, 10-12 núcleos se o tumor for irressecável.[3]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
[40]Lopyan NM, Ehrlich PF. Surgical management of Wilms tumor (nephroblastoma) and renal cell carcinoma in children and young adults. Surg Oncol Clin N Am. 2021 Apr;30(2):305-23.
http://www.ncbi.nlm.nih.gov/pubmed/33706902?tool=bestpractice.com
No protocolo UMBRELLA da SIOP, a biópsia pré-tratamento não é recomendada rotineiramente.[41]Vujanić GM, Gessler M, Ooms AHAG, et al. The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol. 2018 Nov;15(11):693-701.
https://www.nature.com/articles/s41585-018-0100-3
http://www.ncbi.nlm.nih.gov/pubmed/30310143?tool=bestpractice.com
A biópsia percutânea por agulha cortante (biópsia tru-cut) pode ser considerada em pacientes em que se suspeite que o tumor não seja de Wilms, como crianças pequenas com doença em estádio IV e em crianças com >10 anos de idade, pois a frequência de tumores renais que não são de Wilms é maior nessas faixas etárias.[41]Vujanić GM, Gessler M, Ooms AHAG, et al. The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol. 2018 Nov;15(11):693-701.
https://www.nature.com/articles/s41585-018-0100-3
http://www.ncbi.nlm.nih.gov/pubmed/30310143?tool=bestpractice.com
[42]van den Heuvel-Eibrink MM, van Tinteren H, Rehorst H, et al. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP renal tumour study group. Pediatr Blood Cancer. 2011 May;56(5):733-7.
http://www.ncbi.nlm.nih.gov/pubmed/21370404?tool=bestpractice.com
Teste genético
O teste genético molecular é uma ferramenta diagnóstica útil para a identificação de síndromes fenotípicas que podem estar associadas ao tumor de Wilms. Especificamente, os pacientes com tumor de Wilms isolado ou aqueles com tumor de Wilms e anomalias associadas podem ser examinados quanto a deleções de WT1 e outros genes contíguos como PAX6 por hibridização in situ fluorescente (FISH). Os pacientes com características clínicas sugestivas de síndrome de Beckwith-Wiedemann podem ser examinados quanto a uma duplicação de 11p15.5 por análise cromossômica de alta resolução e FISH.[43]Diller L, Ghahremani M, Morgan J, et al. Constitutional WT1 mutations in Wilms tumor patients. J Clin Oncol. 1998 Nov;16(11):3634-40.
http://www.ncbi.nlm.nih.gov/pubmed/9817285?tool=bestpractice.com
Estudos de perda de heterozigosidade (LOH) para 16q e 1p são realizados em tecido tumoral fresco congelado.[44]Wittmann S, Zirn B, Alkassar M, et al. Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes Chromosomes Cancer. 2007 Feb;46(2):163-70.
http://www.ncbi.nlm.nih.gov/pubmed/17099873?tool=bestpractice.com
Em toda criança com tumor de Wilms, deve-se armazenar em um biobanco o material tumoral (tecido tumoral congelado fresco) e tecido renal saudável para realizar estudos de pesquisa que ajudem a definir novos fatores de risco e desenvolver opções de tratamento melhores.