Etiologia
O tumor de Wilms é uma neoplasia geneticamente heterogênea.[3][14] Pode ser hereditário ou ocorrer esporadicamente. Os genes do câncer que estão por trás do tumor de Wilms são diversos e envolvem cerca de 40 genes.[3] A maioria dos genes atua no controle da expressão gênica e na sinalização de fatores de crescimento.[3] Mutações na linha germinativa estão presentes em pelo menos 10% dos pacientes e incluem o primeiro gene implicado na tumorigênese de Wilms, o gene WT1 (tumor 1 de Wilms).[15] Outras mutações na linha germinativa detectadas no artigo da iniciativa TARGET do Children's Oncology Group incluem TP53 (proteína tumoral p53; 17p13), DICER1 (dicer 1, ribonuclease III; 14q32), DIS3L2 (DIS3 semelhante a 3'-5' exoribonuclease 2; 2q37) e algumas variantes inesperadas da linha germinativa que envolvem PALB2 (parceiro e localizador de BRCA2; 16p12) e CHEK2 (quinase de checkpoint 2; 22q12).[15] As mutações somáticas mais prevalentes estão nos genes de processamento WT1 (11p13), CTNNB1 (betacatenina 1; 3p22), AMER1 (proteína de recrutamento de membrana APC 1; Xq11), IGF2 (fator de crescimento semelhante à insulina 2; 11p15), TP53 (17p13), MYCN (proto-oncogene MYCN, fator de transcrição bHLH; 2p24), SIX1 (SIX homeobox 1; 14q23) e SIX2 (SIX homeobox 2; 2p21) e microRNA.[14]
O risco de desenvolver tumor de Wilms é maior em crianças com anomalias congênitas. Em um estudo sobre câncer infantil realizado na Grã-Bretanha, anomalias congênitas foram encontradas em cerca de 8% das crianças com tumor de Wilms.[16] O risco é maior entre crianças com certas síndromes congênitas de supercrescimento, como a síndrome de Perlman, a síndrome de Beckwith-Wiedemann e a síndrome de Simpson-Golabi-Behmel e, também, entre crianças com certas síndromes congênitas que não são de supercrescimento, como a síndrome de Denys-Drash e a síndrome WAGR (tumor de Wilms, aniridia, anormalidades geniturinárias, diversos atrasos no desenvolvimento).[3][17]
Crianças com anomalias geniturinárias congênitas como hipospádia, genitália atípica, rim fundido (em forma de ferradura) ou criptorquidia estão também predispostas a desenvolver tumor de Wilms.[3][16][18]
Estudos têm investigado a associação entre o tumor de Wilms e a exposição pré-natal a fatores ambientais nocivos ou fatores de risco do estilo de vida materno; a maioria deles é inconclusiva ou requer investigações adicionais. No entanto, uma metanálise de estudos tipo de caso-controle identificou uma ligação entre a exposição dos pais a pesticidas durante o período de pré-concepção ou gestação e o aumento do risco de tumor de Wilms.[19]
Fisiopatologia
Muitas mudanças genéticas diferentes convergem em um número limitado de vias de desenvolvimento que resultam na oncogênese do tumor de Wilms.[15] Ao compreender essas vias identificadas, podem-se aplicar possíveis abordagens terapêuticas direcionadas no ambiente clínico.[3]
Acredita-se que a oncogênese do tumor de Wilms seja um processo composto por várias etapas que surge da nefrogênese fetal aberrante, na qual muitos genes relacionados ao tumor de Wilms têm papéis fundamentais no rim em desenvolvimento.[3][14] Como o bloqueio da diferenciação das células progenitoras renais é incompleto, todas as três linhagens do rim em desenvolvimento (blastema, epitélio e estroma) podem ser identificadas na histologia trifásica clássica do tumor de Wilms.[14]
Restos nefrogênicos são focos microscópicos de elementos renais blastemais primitivos encontrados no tecido renal normal em um local intra ou perilobar e são derivados de células-tronco renais. Restos nefrogênicos são encontrados em >90% dos pacientes com tumores de Wilms bilaterais e em cerca de 40% dos pacientes com tumor de Wilms esporádico e unilateral e são considerados lesões precursoras do tumor de Wilms.[3] A aquisição de alterações genéticas e epigenéticas adicionais causa a formação de tumores.
O primeiro modelo genético, ou "hipótese de dois toques" (dois eventos mutacionais limitadores de taxa que ocorrem em sequência), postula que as crianças que desenvolvem tumor de Wilms hereditário nascem com uma mutação constitucional pré-zigótica do ácido desoxirribonucleico (DNA) em um alelo de um gene, que é o evento desencadeante, e que esse evento é seguido por outro evento genético no mesmo locus do gene ou em um locus diferente, causando a formação do tumor.[20][21] O gene WT1 (11p13) e o gene WT2 (11p15) são genes supressores de tumores que desempenham um papel importante no desenvolvimento normal do trato urogenital.[20] A inativação desses genes parece ser um evento genético inicial na evolução do tumor de Wilms. Acredita-se que a perda de heterozigosidade (LOH) em 1p e 16q seja um evento secundário que, em última instância, resulta na formação do tumor de Wilms.[20]
Estudos sugerem que a expressão aberrante do oncogene IGF-2, resultante da perda da impressão do gene IGF-2 materno, pode ser uma das alterações epigenéticas mais comuns do tumor de Wilms.[22] Além disso, restos nefrogênicos intralobares têm maior probabilidade de serem associados à mutação genética idêntica encontrada nos tumores de Wilms originados do mesmo hospedeiro.[23]
Juntos, esses estudos sugerem que os restos nefrogênicos e o tumor podem ser clonalmente derivados de uma célula-tronco renal embrionária comum e que vários eventos genéticos e epigenéticos sequenciais, como a inativação dos genes supressores de tumor e perda de heterozigosidade (LOH) em 1p e 16q, podem ocorrer nessas lesões, causando tumorigênese.[24] Uma análise do tecido histologicamente normal respalda também a hipótese de que as expansões clonais no tecido renal normal, algumas das quais abrigam variantes condutoras, antecedem o desenvolvimento do tumor de Wilms.[25]
Os ganhos de número de cópia na região cromossômica 1q21.1-q31.3 têm se mostrado significativamente associados à recidiva, bem como a desequilíbrios alélicos nos braços cromossômicos 1p, 1q, 3p, 3q e 14q.[26][27]
A análise de pares de tumores primários-recidivantes (em que o DNA está disponível a partir do tumor primário para comparar com o tumor recidivante) sugere que WT2 (11p15) LOH (e outras alterações no número de cópias) e mutações em WT1 e MLLT1 geralmente ocorrem precocemente, mas mutações em SIX1, MYCN e WTX ocorrem mais tarde.[28]
[Figure caption and citation for the preceding image starts]: Deleção do cromossomo 11National Cancer Institute; usado com permissão [Citation ends].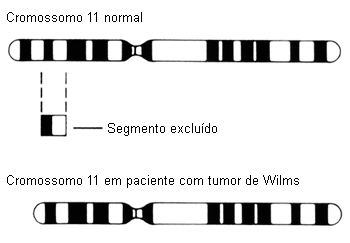
O uso deste conteúdo está sujeito ao nosso aviso legal