Etiologia
A etiologia da hemoglobinúria paroxística noturna (HPN) decorre de dois fatores: uma mutação somática em uma célula-tronco hematopoiética e um histórico de hipoplasia da medula óssea.[4][5] A mutação somática é sempre no gene da âncora fosfatidilinositol glicano (PIG-A), o qual codifica uma enzima necessária para a síntese da âncora de glicosilfosfatidilinositol. Porque a âncora não pode ser sintetizada nas células afetadas, as proteínas ligadas à membrana por esses meios estão ausentes (células HPN III) ou extremamente reduzidas (células HPN II), particularmente as proteínas de defesa do complemento CD55 e CD59.[6] Não se conhece nenhuma outra causa da mutação somática além da taxa natural de mutações decorrente dos erros de transcrição não reparados que ocorrem durante a replicação.[Figure caption and citation for the preceding image starts]: Âncora glicosilfosfatidilinositol na hemoglobinúria paroxística noturna (HPN)Do acervo do Dr. Wendell F. Rosse [Citation ends].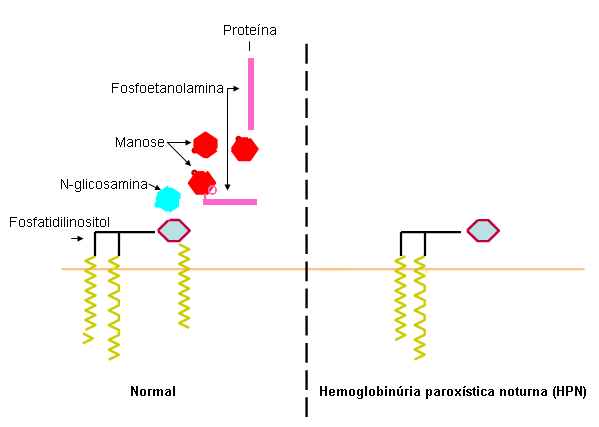
Para que o clone defeituoso aumente suficientemente de tamanho e, consequentemente, cause sintomas clínicos, parece ser necessária a supressão da hematopoiese, como na anemia aplásica.[7] Em muitos casos, acredita-se que isso tenha origem autoimune (ou seja, um ataque mediado por células T à medula óssea), embora hipoplasias induzidas por medicamentos e substâncias químicas também tenham sido observadas. De modo geral, não é possível atribuir uma causa "idiopática" para a hipoplasia da medula. Após a recuperação da medula, o clone deficiente em glicosilfosfatidilinositol pode se tornar dominante. Em aproximadamente 25% dos pacientes, o clone de HPN desaparecerá com terapia imunossupressora.[8] Os pacientes restantes manterão um clone. Em 30% dos pacientes, o clone irá expandir e os pacientes poderão desenvolver sintomas clínicos.[8]
Fisiopatologia
Muito da fisiopatologia hemoglobinúria paroxística noturna (HPN) clássica surge dos efeitos da ativação do complemento nas células sanguíneas que encontram-se sem as proteínas de defesa do complemento CD55 e, especialmente, CD59.[1] Assim, mesmo uma pequena ativação que normalmente ocorre o tempo todo (causando uma hemólise de baixo grau contínua) e é exagerada à noite (causando exacerbações noturnas) resulta no depósito de mais complexos terminais líticos nas células sanguíneas anormais. Para os eritrócitos, isso resulta na penetração da membrana e em lise celular intravascular, provocando a liberação de hemoglobina no plasma que, se suficiente, causará hemoglobinúria. O complemento pode ser ainda mais ativado também por infecções, cirurgias e traumas, resultando em paroxismos de hemólise; embora os paroxismos também podem ocorrer sem causa aparente. A quantidade de hemólise depende do número de eritrócitos anormais (tamanho do clone), de sua anormalidade e do grau de ativação do complemento.
A hemoglobinemia resultante rapidamente liga o óxido nítrico aos tecidos, resultando na contração do músculo liso que se manifesta por meio de espasmo esofágico e dor abdominal.[9] Essa privação de óxido nítrico pode causar espasmo vascular, resultando em disfunção erétil, hipertensão pulmonar e disfunção renal. A maioria dos pacientes apresenta uma "fadiga" incomum que é exacerbada durante os episódios hemoglobinúricos. A hemoglobinúria pode afetar a função renal agudamente, quando maciça, ou cronicamente causando uma insuficiência renal potencial.[10]
A ativação do complemento nas plaquetas anormais resulta em sua ativação e isso, junto com outras causas, resulta em trombose originalmente venosa, mas eventos arteriais também podem ocorrer.[1][11] Isso frequentemente ocorre em locais incomuns, como as veias hepáticas e abdominais ou os seios venosos cerebrais, mas também são comuns nas veias profundas e ocasionalmente nas artérias.
Classificação
Classificação proposta[3]
Hemoglobinúria paroxística noturna (HPN) hemolítica clássica: tamanho do clone grande o suficiente para resultar em evidência clínica de hemólise sem evidência de outro distúrbio da medula óssea
HPN no cenário de outro distúrbio da medula óssea, como a anemia aplásica associada a HPN (evidência de HPN no cenário de anemia aplásica) e a síndrome mielodisplásica associada a HPN de baixo risco (evidência de HPN no cenário de mielodisplasia)
HPN subclínica: tamanho do clone muito pequeno para resultar em evidência de hemólise
O uso deste conteúdo está sujeito ao nosso aviso legal