Abordagem
Um diagnóstico preciso requer uma descrição detalhada dos episódios clínicos e análise cuidadosa do eletroencefalograma (EEG).
História
É fundamental obter uma descrição detalhada de uma testemunha ocular para todas as epilepsias. Gravações domésticas em vídeo podem ter enorme valor, e a observação direta das convulsões no ambiente clínico pode ser possível, pois as salvas tendem a ocorrer com frequência ao longo do dia.
Os espasmos são geralmente simétricos, bilaterais, com contrações breves dos grupos musculares axiais e podem afetar os flexores, extensores ou ambos. O espasmo flexor é o mais comum; no entanto, a manifestação pode ser sutil, com um simples movimento vertical breve da cabeça ou vibrações na pálpebra. No início da evolução da doença, os espasmos podem ser eventos isolados com a tendência de desenvolvimento de salvas posteriores. Salvas de até 50-100 espasmos seriais podem ocorrer frequentemente em 24 horas. Os espasmos podem ser desencadeados no início/fim do sono ou pela alimentação, frequentemente induzindo ao diagnóstico equivocado de refluxo gastroesofágico ou cólica infantil.
A idade típica de início é entre 1 mês e 1 ano de idade, com idade pico de início entre 3 e 5 meses. Em alguns, a manifestação pode ocorrer até os 3 anos. Pode haver história pregressa de atraso no desenvolvimento, o qual pode ser consistente com a etiologia subjacente (por exemplo, síndrome de Down). Entretanto, o início dos espasmos está frequentemente associado com a estabilização em um patamar ou a regressão do desenvolvimento, até mesmo em lactentes previamente considerados como tendo desenvolvimento normal. Na apresentação, deve-se dar atenção cuidadosa ao estado de desenvolvimento passado e atual da criança.
Uma história detalhada pré-natal e do parto pode sugerir lesões perinatais significativas com impacto no desenvolvimento cerebral, como hipóxia, acidente vascular cerebral (AVC) pré-natal/neonatal ou trombose sinovenosa. Soluços, dificuldades de alimentação com resposta de sobressalto exagerada e retardo do crescimento pôndero-estatural podem indicar uma etiologia metabólica potencial. Deve-se dar atenção cuidadosa à história familiar, que pode sugerir uma etiologia subjacente (por exemplo, esclerose tuberosa). Normalmente, há história familiar de outras síndromes epilépticas.
Exame
Um exame físico geral cuidadoso deve identificar a dismorfia e os estigmas neurocutâneos associados com síndromes genéticas comuns, como a síndrome de Down e a esclerose tuberosa. O exame físico com a lâmpada de Wood é essencial, pois as máculas hipomelanóticas (em formato de folha) típicas da esclerose tuberosa podem não ser observadas na sua ausência. As lesões ficam fluorescentes sob a luz ultravioleta.[Figure caption and citation for the preceding image starts]: Máculas hipomelanóticas (em formato de folha) em uma criança com esclerose tuberosaDo acervo da Dra. Teesta Soman e do Dr. Shelly Weiss [Citation ends].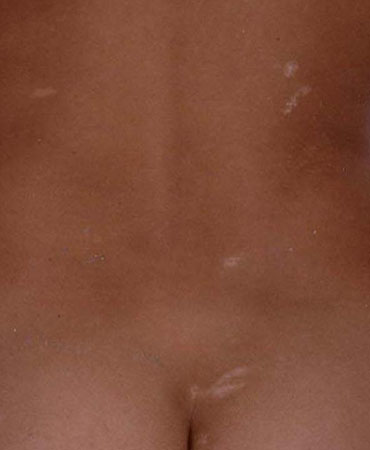
Os parâmetros de crescimento, incluindo perímetro cefálico, devem ser anotados precisamente em tabelas apropriadas. A microcefalia na apresentação pode sugerir malformações cerebrais estruturais, lesões perinatais, eventos vasculares, distúrbios metabólicos hereditários ou infecções intrauterinas.
O exame físico de tônus e os padrões de movimento podem revelar hipotonia troncular ou espasticidade periférica. Assimetria ou deficit focal no exame físico podem sugerir malformações corticais, lesões com efeito de massa, infecção ou lesão precedente no cérebro em desenvolvimento. Entretanto, na apresentação, o exame físico neurológico inicial pode ser normal e fornecer poucas informações.
Um exame físico cardiovascular pode identificar comorbidades de esclerose tuberosa ou síndromes genéticas como a síndrome de Down. O exame oftalmológico pode sugerir uma síndrome subjacente ou infecção perinatal (por exemplo, citomegalovírus [CMV] ou toxoplasmose).
É importante registrar o estado de desenvolvimento pré-mórbido na apresentação, pois o lactente pode possuir desenvolvimento adequado para a idade ou características de atraso no desenvolvimento. O atraso pode ocorrer em todas as áreas de desenvolvimento, incluindo coordenação motora fina, coordenação motora grossa, fala e linguagem e visão.
eletroencefalograma (EEG)
Deve-se realizar um EEG em todos pacientes assim que houver suspeita do diagnóstico. A diretriz do National Institute for Health and Care Excellence (NICE) do Reino Unido recomenda que crianças menores de 2 anos de idade com espasmos infantis suspeitos ou confirmados sejam encaminhadas a um neurologista pediátrico especialista dentro de 24 horas para garantir uma avaliação rápida, incluindo um EEG do sono.[26] Em geral, o EEG revela hipsarritmia, embora não de maneira universal.[27]
[Figure caption and citation for the preceding image starts]: Eletroencefalograma (EEG) demonstrando hipsarritmiaDo acervo da Dra. Teesta Soman e do Dr. Shelly Weiss [Citation ends].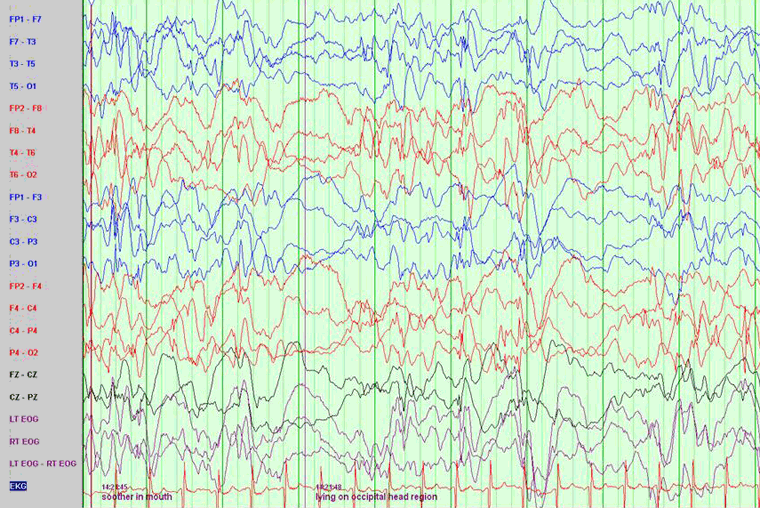
A hipsarritmia é caracterizada por uma espícula aleatória de alta voltagem e ondas lentas de amplitude variável, surgindo de focos múltiplos que variam com o tempo. A atividade de fundo é assíncrona e geralmente caótica.[28] Há maior probabilidade de hipsarritmia típica nas fases precoces dos espasmos infantis e em pacientes mais jovens.[27] Ela pode evoluir com o tempo para descargas epileptiformes interictais e multifocais. O EEG interictal é muito anormal nessa síndrome, mas ocasionalmente é apenas anormal durante o sono. A hipsarritmia pode desaparecer durante o sono REM e é observada com maior sensibilidade durante outros estágios do sono.
Se há forte suspeita clínica de espasmos infantis, é necessário um registro de sono prolongado, pois o padrão hipsarrítmico típico pode não ser percebido.[27]
A hipsarritmia pode ser assimétrica, sugerindo epilepsia lesional, malformação cortical ou distúrbio de migração neuronal. A hipsarritmia modificada pode ser mais sincrônica, menos simétrica e mostrar mais alterações focais ou áreas de atenuação. Descargas focais interictais (multi) também podem ser observadas no EEG.
O EEG ictal é variável, mas pode mostrar supressão, atividade rápida ou atenuação da atividade de fundo (uma resposta eletrodecremental), que pode ser precedida por ondas lentas de vértice de alta amplitude ou atividade tipo fusiforme.
Exames por imagem
Uma ressonância nuclear magnética (RNM) cranioencefálica pode identificar anormalidades migratórias e estruturais subjacentes. O exame pode mostrar áreas de malformação ou lesões secundárias a hemorragia, infecção, infarto ou tumor (por exemplo, calcificação, porencefalia [cisto ou cavidade no hemisfério cerebral] e encefalomalácia [amolecimento do tecido cerebral]). Sinais de esclerose tuberosa podem ser observados, incluindo hamartomas corticais, nódulos subependimários e astrocitomas subependimários de células gigantes. Padrões anormais característicos podem sugerir síndromes neurocutâneas ou neurometabólicas. Entretanto, os exames de neuroimagem podem ser inteiramente normais.
Uma tomografia computadorizada (TC) cranioencefálica é menos sensível e deve ser considerada apenas se a RNM não for inicialmente de fácil obtenção.[Figure caption and citation for the preceding image starts]: Tubérculos corticais na ressonância nuclear magnética (RNM) na esclerose tuberosaDo acervo da Dra. Teesta Soman e do Dr. Shelly Weiss [Citation ends].
Teste genético
Se disponível, e se nenhuma etiologia subjacente clara for encontrada após exame clínico e RNM, testes genéticos de sequenciamento de próxima geração (painel de genes/sequenciamento de exoma total/sequenciamento de genoma completo) devem ser considerados precocemente.[29][30][31] O sequenciamento de próxima geração também deve ser considerado para pacientes com distúrbios cerebrais estruturais conhecidos por estarem associados a mutações genéticas, como a esclerose tuberosa. A análise de hibridização genômica comparativa (CGH) por microarray também é recomendada na maioria dos pacientes, pois anormalidades cromossômicas e variantes no número de cópias foram associadas a espasmos infantis. Se a CGH por microarray não estiver disponível, deve proceder-se a análise cromossômica.
Uma etiologia genética pode ser definida em até 41% dos casos de espasmos infantis, incluindo trissomia do cromossomo 21, CDKL5, STXBP1, ARX, IQSEC2, TSC1, TSC2 e mais.[31][32][33] Uma mutação genética pode ser de novo ou herdada de um pai com sintomas leves ou de um pai não afetado. Os testes genéticos de sequenciamento de próxima geração são preferencialmente realizados como uma análise em trio, o que significa que as amostras dos pais biológicos e das crianças são coletadas e analisadas simultaneamente.
Investigações metabólicas e outras investigações laboratoriais
Além dos testes genéticos, os testes iniciais na apresentação devem incluir:
Hemograma completo
Ureia e eletrólitos
Glicose
Cálcio e magnésio séricos
Testes de função hepática
Amônia sérica
Gasometria/bicarbonato
Lactato sérico/piruvato
Aminoácidos plasmáticos
Biotinidase
Ácidos orgânicos urinários
Acilcarnitinas
Creatina e guanidinoacetato na urina e no plasma
Alfa-aminoadípica semialdeído desidrogenase (AASA) na urina
Exame do líquido cefalorraquidiano (LCR): deve incluir glicose e lactato no LCR pareado com plasma, aminoácidos, piridoxal 5-fosfato e metiltetraidrofolato.
Posteriormente, os seguintes exames podem ser considerados:
Testes de função tireoidiana (tiroxina livre [T4L], hormônio estimulante da tireoide [TSH])
Glicoformas de transferrina plasmática
Ácidos graxos de cadeia muito longa
Sulfocisteína na urina.
Outros exames a serem considerados, se indicados:
Um rastreamento para infecções intrauterinas, incluindo citomegalovírus (CMV) e toxoplasmose (anticorpos contra toxoplasma e CMV, e reação em cadeia da polimerase para CMV na urina)
Ecocardiografia e ultrassonografia renal se houver evidências clínicas ou radiológicas de esclerose tuberosa
Revisão oftalmológica para evidências de coriorretinite em infecções intrauterinas
O uso deste conteúdo está sujeito ao nosso aviso legal