Etiologia
As células de Langerhans são células dendríticas epidérmicas que apresentam antígeno contra e ativam linfócitos T específicos do antígeno. Elas são encontradas na maioria dos órgãos. A HCL é considerada uma neoplasia mieloide inflamatória. A condição é causada por uma proliferação clonal de células precursoras mieloides, que se diferenciam em células de Langerhans patológicas nas lesões.[13][14]
Mutações ativadoras na via da proteína quinase ativada por mitógenos (MAPK), em particular a BRAF (proto-oncogene B-Raf) V600E, foram identificadas em 50% a 60% dos casos de HCL.[15][16][17] A BRAF é uma tirosina quinase citoplasmática envolvida na transdução do sinal do fator de crescimento. Mutações ativadoras, como a BRAF V600E, causam o aumento da transcrição de BCL-2 como 1 (BCL2L1), que inibe a apoptose, e diminuição da transcrição do receptor de quimiocina do tipo C-C 7 (CCR7), que estimula a maturação de células dendríticas e estimula a mobilização de células de Langerhans ativadas para a drenagem dos linfonodos.[3][18][19] O acúmulo resultante de células dendríticas patológicas contribui para a inflamação local e sistêmica e pode se infiltrar ou danificar o tecido circundante.[3][20]
A ativação de mutações na via MAPK pode ocorrer em diferentes estágios da diferenciação mieloide. Acredita-se que as mutações em células-tronco hematopoiéticas pluripotentes (que expressam CD34) causem HCL de alto risco, enquanto as mutações em precursores de células dendríticas e monócitos (que expressam CD11c ou CD14) deem origem à HCL de baixo risco.[13][16]
Fisiopatologia
A HCL é caracterizada por lesões granulomatosas que contêm proliferação clonal de células de Langerhans patológicas, que expressam CD1a, CD207 (langerina) e S100. As células HCL são grandes, com um núcleo em forma de rim.[14][Figure caption and citation for the preceding image starts]: Micrografia de ampliação muito alta de histiocitose das células de Langerhans. Coloração H&E. Caracteriza-se por histiócitos do tipo Langerhans que possuem núcleo reniforme (em forma de rim) e coram com S100 e CD1aNephron. Reproduzido sob uma licença Creative Commons CC BY-SA 3.0: https://creativecommons.org/licenses/by-sa/3.0/deed.en [Citation ends].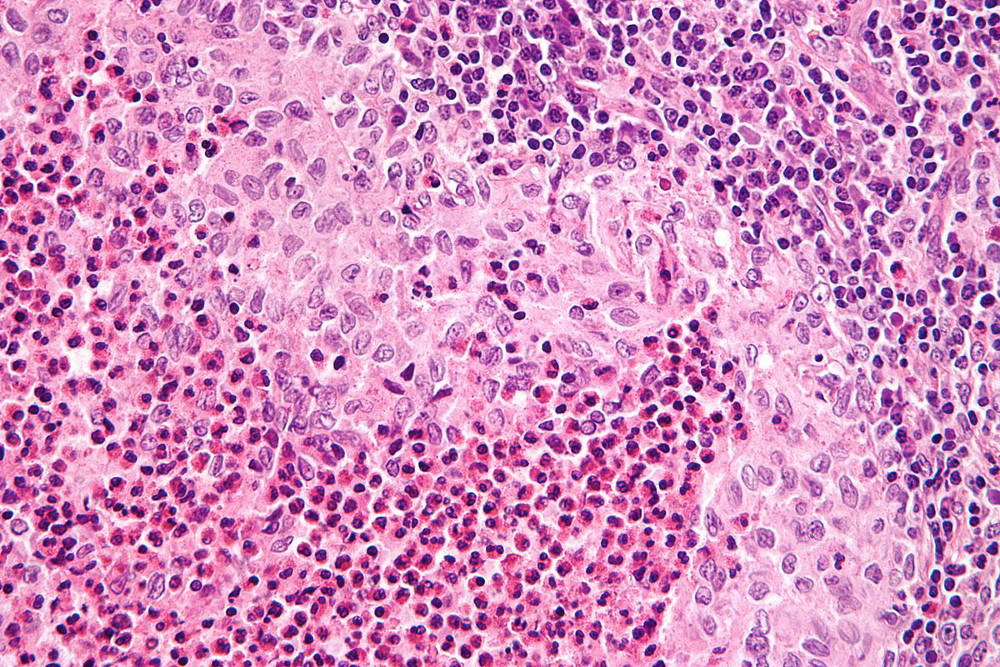
As células neoplásicas de Langerhans ativam e recrutam outras células inflamatórias, o que causa o desenvolvimento de um infiltrado inflamatório que inclui células T ativadas, eosinófilos, neutrófilos e macrófagos.[14] Desenvolve-se desregulação imunológica local, caracterizada pelo aumento da expressão de citocinas pró-inflamatórias, remodelação tecidual e neoangiogênese.[21] Fatores ambientais, como tabagismo ou infecção, contribuem para o desenvolvimento e a persistência da inflamação.[20][22] O tabagismo causa acúmulo de células de Langerhans nos pulmões e induz a produção de citocinas pró-inflamatórias, o que causa formação de granuloma histiocítico.[20]
Classificação
Histiocyte Society: classificação de histiocitose[1]
Grupo L (Langerhans)
Histiocitose das células de Langerhans (HCL)
HCL em sistema único
HCL pulmonar positiva
HCL positiva para vários órgãos (VO) e órgãos de risco (OR)
HCL negativa para VO e RO
Associada a outro distúrbio mieloproliferativo/mielodisplásico
Histiocitose celular indeterminada
Doença de Erdheim Chester (DEC) tipo clássico
DEC sem comprometimento ósseo
DEC associada a outro distúrbio mieloproliferativo/mielodisplásico
Xantogranuloma juvenil extracutâneo ou disseminado com translocações ativadoras de proteína quinase ativada por mitógenos (MAPK) ou quinase do linfoma anaplásico (ALK)
DEC e HCL mistas
Grupo C (cutâneo)
Histiocitoses cutâneas não HCL
Família do xantogranuloma
Família não xantogranuloma
Histiocitoses cutâneas não HCL com um importante componente sistêmico
Xantoma disseminado
Reticulocitose multicêntrica
Grupo M (maligno)
Histiocitose maligna primária
Histiocitose maligna secundária
Grupo R (doença de Rosai-Dorfman e histiocitoses não cutâneas diversas de células não Langerhans)
Doença de Rosai-Dorfman (DRD) familiar
DRD clássica (nodal)
DRD extranodal
DRD associada a neoplasia
DRD associada a doença imunológica
Grupo H (linfo-histiocitose hemofagocítica e síndrome de ativação de macrófagos)
Linfo-histiocitose hemofagocítica (LHH) primária
LHH secundária
Rede Euro Histio: classificação clínica de HCL pediátrica[2]
Sistema único:
Um sistema de órgãos comprometido
Geralmente ossos, pele, linfonodos, pulmões ou tireoide ou hipófise.
Sistema múltiplo:
2 ou mais sistemas de órgãos comprometidos
Pode ser com ou sem comprometimento de órgãos de risco.
Órgãos de risco:
Determinados órgãos são considerados de alto risco devido às elevadas taxas de mortalidade associadas nos pacientes que não respondem ao tratamento
Os órgãos de alto risco incluem fígado, baço e medula óssea.
Histiocyte Society: classificação clínica da HCL em adultos[4]
HCL unifocal: lesão solitária envolvendo qualquer órgão
HCL pulmonar de sistema único: envolvimento pulmonar isolado (predominantemente relacionado ao tabagismo)
HCL multifocal de sistema único: >1 lesão envolvendo qualquer órgão
HCL multissistêmica: envolvimento de ≥2 órgãos/sistemas.
O uso deste conteúdo está sujeito ao nosso aviso legal