História e exame físico
Principais fatores diagnósticos
comuns
presença de fatores de risco
Os principais fatores de risco incluem gamopatia monoclonal de significado indeterminado (MGUS), doenças inflamatórias, infecções crônicas e história familiar positiva.
estase jugular
Uma pressão de enchimento alta do lado direito produz níveis elevados de distensão venosa jugular.[74]
edema nos membros inferiores
Deve-se à hipoalbuminemia decorrente de síndrome nefrótica, e pode decorrer de pressões de enchimento altas do lado direito na presença de cardiomiopatia restritiva. Presente em aproximadamente 50% dos pacientes.[73]
Incomuns
história de gamopatia monoclonal de significado indeterminado (MGUS)
A história pode revelar um diagnóstico prévio de MGUS.[58]
Pacientes com MGUS têm um risco relativo de progressão para amiloidose do tipo AL (o tipo mais comum de amiloidose) de oito a nove vezes.[4][58][59]
É importante observar, entretanto, que uma gamopatia monoclonal incidental pode estar presente em pacientes com outros tipos de amiloidose (por exemplo, amiloidose hereditária, amiloidose por transtirretina do tipo selvagem [ATTRwt]), o que pode levar a um diagnóstico incorreto de amiloidose do tipo AL.[28][29][61]
história de uma condição inflamatória crônica, infecção crônica, síndrome febril periódica hereditária
A amiloidose secundária (AA) é tipicamente associada a: condições inflamatórias crônicas (por exemplo, poliartropatia inflamatória, doença inflamatória intestinal [especificamente doença de Crohn]); infecções crônicas (por exemplo, bronquiectasia, tuberculose, injeção subcutânea de drogas ilícitas, úlceras de decúbito, infecções crônicas do trato urinário, osteomielite); síndrome febril periódica hereditária (por exemplo, febre familiar do Mediterrâneo, síndrome periódica associada ao receptor do fator de necrose tumoral (TNF) [TRAPS], síndromes periódicas associadas à criopirina [CAPS; como a síndrome de Muckle-Wells], deficiência de mevalonato-quinase [anteriormente conhecida como síndrome de hiperimunoglobulinemia D]).[16][24][25][26][27]
púrpura periorbital
A púrpura amiloide ocorre em aproximadamente 15% dos pacientes com amiloidose do tipo AL.[49] Normalmente, é periorbital, mas pode ocorrer em qualquer lugar acima da linha mamilar.[Figure caption and citation for the preceding image starts]: Equimose periorbital bilateral (púrpura amiloide) em paciente com amiloidose do tipo ALWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends]. [Figure caption and citation for the preceding image starts]: Púrpura periorbital clássicaMorie A. Gertz, MD; cortesia da Mayo Clinic [Citation ends].
[Figure caption and citation for the preceding image starts]: Púrpura periorbital clássicaMorie A. Gertz, MD; cortesia da Mayo Clinic [Citation ends].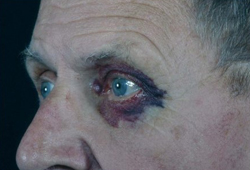
petéquias palpebrais
Petéquias nas pálpebras são comuns; só são evidentes quando os olhos estão fechados. Podem ser confundidas com trombocitopenia imune (PTI) ou coagulopatia, mas são altamente específicas para amiloidose.
macroglossia
Sinal específico e diagnóstico de amiloidose do tipo AL.[49] Ocorre em aproximadamente 10% dos pacientes, mas não é facilmente percebida, pois a apresentação mais comum são as endentações dentárias na parte de baixo da língua.[Figure caption and citation for the preceding image starts]: Macroglossia em paciente com amiloidose do tipo ALWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends].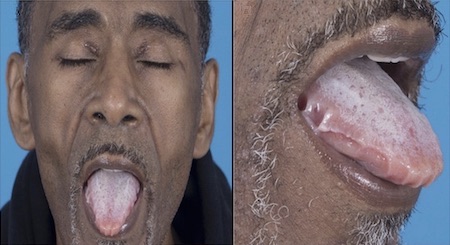
Outros fatores diagnósticos
comuns
fadiga
Geralmente, existem manifestações da doença sistêmica presentes em pacientes com cardiomiopatia amiloide e síndrome nefrótica precoces. Presente em aproximadamente 60% dos pacientes.[42]
perda de peso
Perda extrema de peso (por exemplo, >9 kg) é comum (particularmente em pacientes com envolvimento cardíaco e hepático) e é sugestiva de amiloidose, se associada a edema ou neuropatia.[49]
Uma anamnese pode exigir uma busca detalhada por neoplasias ocultas.
dispneia ao esforço
A dispneia ao esforço é um achado comum em pacientes com cardiomiopatia amiloide. Associada a edema quando as pressões de enchimento no lado direito estão elevadas. Presente em aproximadamente 40% dos pacientes.[49]
Incomuns
neuropatia periférica
O envolvimento nervoso pode levar à neuropatia periférica e é mais comumente associado à amiloidose de cadeia leve de imunoglobulina (AL) e à amiloidose por transtirretina hereditária (ATTRv). A neuropatia periférica leve pode ocorrer em pacientes com amiloidose por transtirretina do tipo selvagem (ATTRwt) (até 20%).[56]
O envolvimento nervoso não é uma característica típica da amiloidose secundária (AA), amiloidose hereditária não associada à TTR ou amiloidose por LECT2.[3]
Geralmente, a apresentação inicial é a perda sensorial distal simétrica (ou seja, perda da percepção de temperatura e da dor, seguida de perda proprioceptiva).[51] Os pacientes geralmente relatam disestesia e parestesia dos pés e pernas, que progridem para as mãos e braços ao longo do tempo.
A neuropatia periférica é uma pista diagnóstica importante para a amiloidose do tipo AL e para a amiloidose por ATTRv.[51]
neuropatia autonômica
O envolvimento nervoso pode levar à neuropatia autonômica e é mais comumente associado à amiloidose do tipo AL e à amiloidose por ATTRv.
O envolvimento nervoso não é uma característica típica da amiloidose AA, amiloidose hereditária não associada à TTR ou amiloidose por LECT2.[3]
Pacientes com neuropatia autonômica podem apresentar disfunção erétil, hipotensão ortostática, disfunção gastrointestinal ou disfunção urinária.[19][69] Anormalidades de sudorese e insuficiência da frequência cardíaca ao mudar a posição corporal são sinais de disfunção autonômica.
A neuropatia autonômica é uma pista diagnóstica importante para a amiloidose do tipo AL e para a amiloidose por ATTRv.[51]
claudicação
Consequência do comprometimento amiloide de pequenos vasos nas artérias periféricas. Resulta em claudicação da mandíbula, panturrilha e membros. Raramente, os pacientes podem apresentar angina.
náuseas ou vômitos
Se o trato gastrointestinal superior estiver envolvido, náuseas ou vômitos podem estar presentes (sintomas pseudo-obstrutivos).
cólicas abdominais
Se o trato gastrointestinal superior estiver envolvido, podem ocorrer cólicas abdominais pós-prandiais (sintomas de pseudo-obstrução).
hábito intestinal alternado
Pode haver incontinência fecal grave alternando com 3-4 dias de constipação.
Esteatorreia
Sinal típico de envolvimento intestinal.
tontura
A tontura pode ser consequência de amiloidose cardíaca (baixo débito cardíaco com fração de ejeção preservada na ecocardiografia) ou síndrome nefrótica (hipoalbuminemia e contração do volume intravascular).
aumento da glândula salivar submandibular
Específico para amiloidose do tipo AL. Pode ser mal interpretado como linfadenopatia. O comprometimento da glândula salivar resulta em uma síndrome seca. Esses pacientes muitas vezes são diagnosticados erroneamente como tendo síndrome de Sjögren.
hepatomegalia
Hepatomegalia palpável >5 cm abaixo da margem costal direita, relatada com mais frequência na amiloidose do tipo AL (aproximadamente 10% dos pacientes).[16][49]
A hepatomegalia palpável é rara na amiloidose AA, mas alterações histopatológicas no fígado podem ser evidentes. O comprometimento hepático é raro em pacientes com amiloidose hereditária.
ombros com aspecto de ombreira (sinal de "shoulder pad")
Um sinal raro e específico da amiloidose do tipo AL. A infiltração periarticular com amiloide produz pseudo-hipertrofia, resultando no aumento da musculatura das cinturas escapular e pélvica.
fraqueza muscular difusa
Pode ocorrer em decorrência de infiltração extracelular de amiloide no músculo (hipertrofia muscular) ou de oclusão vascular, que causa isquemia muscular e claudicação (atrofia muscular).
hipotensão ortostática
Hipotensão ortostática com síncope pode ocorrer se houver neuropatia autonômica (por exemplo, na amiloidose do tipo AL ou amiloidose por ATTRv).
síndrome do túnel do carpo
A prevalência da síndrome do túnel do carpo é maior em pacientes com amiloidose cardíaca por TTR (20.3% vs. 4.1% na população em geral), mas também é uma manifestação da amiloidose do tipo AL.[70]
Deve-se pesquisar o sinal de Tinel (a percussão sobre o nervo do carpo no punho produz formigamento nos dedos polegar, indicador e médio) e realizar a manobra de Phalen (o ato de manter a superfície dorsal das mãos unidas em flexão forçada por cerca de 1 minuto resulta em formigamento nos dedos polegar, indicador e médio) para examinar se há comprometimento pela síndrome do túnel do carpo nos pacientes que se queixarem de parestesia nas mãos.
distúrbios musculoesqueléticos
Fatores de risco
Fortes
gamopatia monoclonal de significado indeterminado (MGUS)
poliartropatia inflamatória
Causa subjacente mais comum de amiloidose secundária (AA).
Inclui pacientes com artrite reumatoide, artrite juvenil, artrite psoriática e espondilite anquilosante.[16]
infecções crônicas
Risco de amiloidose secundária (AA).
As causas incluem bronquiectasias, tuberculose, injeção subcutânea de drogas ilícitas, úlceras de decúbito, infecções do trato urinário crônicas e osteomielite.[16]
doença inflamatória intestinal
Risco de amiloidose secundária (AA).
Em particular, doença de Crohn.[16]
síndromes febris periódicas hereditárias
Febre familiar do Mediterrâneo, síndrome periódica associada ao receptor do fator de necrose tumoral (TNF) (TRAPS), síndromes periódicas associadas à criopirina (CAPS; por exemplo, síndrome de Muckle-Wells) e deficiência de mevalonato-quinase (antes conhecida como síndrome de hiperimunoglobulinemia D) foram implicadas na amiloidose secundária (AA).[24][25][26][27]
O uso deste conteúdo está sujeito ao nosso aviso legal