A investigação para diagnóstico da amiloidose inclui história detalhada e exame físico; avaliação laboratorial e patológica (incluindo estudos para confirmar a presença e o tipo de depósitos amiloides no tecido); e estudos de imagem.
É importante determinar o tipo de amiloidose ao fazer um diagnóstico, pois isso orienta o tratamento.
História
Uma história detalhada deve ser realizada para ajudar a determinar a causa potencial e o tipo de amiloidose.
A história pode revelar um diagnóstico prévio de gamopatia monoclonal de significado indeterminado (MGUS).[58]Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002 Feb 21;346(8):564-9.
https://www.doi.org/10.1056/NEJMoa01133202
http://www.ncbi.nlm.nih.gov/pubmed/11856795?tool=bestpractice.com
Pacientes com MGUS têm um risco relativo de progressão para amiloidose do tipo AL (o tipo mais comum de amiloidose) de oito a nove vezes.[4]Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016 Jun 25;387(10038):2641-54.
http://www.ncbi.nlm.nih.gov/pubmed/26719234?tool=bestpractice.com
[58]Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002 Feb 21;346(8):564-9.
https://www.doi.org/10.1056/NEJMoa01133202
http://www.ncbi.nlm.nih.gov/pubmed/11856795?tool=bestpractice.com
[59]Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018 Jan 18;378(3):241-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5852672
http://www.ncbi.nlm.nih.gov/pubmed/29342381?tool=bestpractice.com
É importante observar, entretanto, que uma gamopatia monoclonal incidental pode estar presente em pacientes com outros tipos de amiloidose (por exemplo, amiloidose hereditária, amiloidose por transtirretina do tipo selvagem [ATTRwt]), o que pode levar a um diagnóstico incorreto de amiloidose do tipo AL.[28]Comenzo RL, Zhou P, Fleisher M, et al. Seeking confidence in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood. 2006 May 1;107(9):3489-91.
http://www.bloodjournal.org/content/107/9/3489.full
http://www.ncbi.nlm.nih.gov/pubmed/16439680?tool=bestpractice.com
[29]Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002 Jun 6;346(23):1786-91.
http://www.nejm.org/doi/full/10.1056/NEJMoa013354#t=article
http://www.ncbi.nlm.nih.gov/pubmed/12050338?tool=bestpractice.com
[61]Phull P, Sanchorawala V, Connors LH, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018 Mar;25(1):62-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6157907
http://www.ncbi.nlm.nih.gov/pubmed/29424556?tool=bestpractice.com
A conscientização clínica, juntamente com uma história familiar cuidadosa e avaliação laboratorial/patológica (incluindo testes genéticos) são essenciais para evitar um diagnóstico incorreto.[30]Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020 Sep 23;21(1):198.
https://www.doi.org/10.1186/s12875-020-01252-4
http://www.ncbi.nlm.nih.gov/pubmed/32967612?tool=bestpractice.com
[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
A amiloidose secundária (AA) está associada a:[16]Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun 7;356(23):2361-71.
https://www.nejm.org/doi/10.1056/NEJMoa070265
http://www.ncbi.nlm.nih.gov/pubmed/17554117?tool=bestpractice.com
[22]Fayand A, Boutboul D, Galicier L, et al. Epidemiology of Castleman disease associated with AA amyloidosis: description of 2 new cases and literature review. Amyloid. 2019 Dec;26(4):197-202.
http://www.ncbi.nlm.nih.gov/pubmed/31364863?tool=bestpractice.com
[23]Bernabei L, Waxman A, Caponetti G, et al. AA amyloidosis associated with Castleman disease: a case report and review of the literature. Medicine (Baltimore). 2020 Feb;99(6):e18978.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7015640
http://www.ncbi.nlm.nih.gov/pubmed/32028407?tool=bestpractice.com
[24]Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017 Aug;31(4):596-609.
http://www.ncbi.nlm.nih.gov/pubmed/29773275?tool=bestpractice.com
[25]Delaleu J, Deshayes S, Rodrigues F, et al. Tumour necrosis factor receptor-1 associated periodic syndrome (TRAPS)-related AA amyloidosis: a national case series and systematic review. Rheumatology (Oxford). 2021 Dec 1;60(12):5775-84.
https://academic.oup.com/rheumatology/article/60/12/5775/6170651
http://www.ncbi.nlm.nih.gov/pubmed/33715002?tool=bestpractice.com
[26]Rodrigues F, Cuisset L, Cador-Rousseau B, et al. AA amyloidosis complicating cryopyrin-associated periodic syndrome: a study of 86 cases including 23 French patients and systematic review. Rheumatology (Oxford). 2022 Nov 28;61(12):4827-34.
http://www.ncbi.nlm.nih.gov/pubmed/35262642?tool=bestpractice.com
[27]Lachmann HJ, Goodman HJ, Andrews PA, et al. AA amyloidosis complicating hyperimmunoglobulinemia D with periodic fever syndrome: a report of two cases. Arthritis Rheum. 2006 Jun;54(6):2010-4.
https://onlinelibrary.wiley.com/doi/epdf/10.1002/art.21901
http://www.ncbi.nlm.nih.gov/pubmed/16732551?tool=bestpractice.com
condições inflamatórias crônicas (por exemplo, poliartropatia inflamatória, doença inflamatória intestinal [especificamente doença de Crohn])
infecções crônicas (por exemplo, bronquiectasia, tuberculose, injeção subcutânea de drogas ilícitas, úlceras de decúbito, infecções crônicas do trato urinário, osteomielite)
síndrome febril periódica hereditária (por exemplo, febre familiar do Mediterrâneo, síndrome periódica associada ao receptor do fator de necrose tumoral [TNF] [TRAPS], síndromes periódicas associadas à criopirina [CAPS; como a síndrome de Muckle-Wells], deficiência de mevalonato-quinase [anteriormente conhecida como síndrome de hiperimunoglobulinemia D])
Doença de Castleman (variante plasmocitária), um distúrbio linfoproliferativo não canceroso, mas é raro.
A ATTRwt está associada ao envelhecimento e afeta principalmente homens idosos. A história geralmente inclui cardiomiopatia, síndrome do túnel do carpo e estenose da coluna vertebral.[63]Law S, Gillmore JD. When to suspect and how to approach a diagnosis of amyloidosis. Am J Med. 2022 Apr;135 Suppl 1:S2-8.
https://www.amjmed.com/article/S0002-9343(22)00029-8/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35081377?tool=bestpractice.com
Sintomas
Pacientes com amiloidose frequentemente apresentam sintomas relacionados a uma síndrome clínica do órgão afetado (por exemplo, cardiomiopatia, síndrome nefrótica, neuropatia).[64]Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020 Dec 3;136(23):2620-7.
https://www.sciencedirect.com/science/article/pii/S0006497120819582
http://www.ncbi.nlm.nih.gov/pubmed/33270858?tool=bestpractice.com
Fadiga, perda de peso, parestesias e dispneia ao esforço são os sintomas mais comuns associados à amiloidose e são comuns a todas as formas sistêmicas.[42]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis: diagnosis and management. Clin Lymphoma Myeloma. 2005 Nov;6(3):208-19.
http://www.ncbi.nlm.nih.gov/pubmed/16354326?tool=bestpractice.com
No entanto, essas queixas são inespecíficas.
Perda extrema de peso (por exemplo, >9 kg) é comum (particularmente em pacientes com envolvimento cardíaco e hepático) e é sugestiva de amiloidose, se associada a edema ou neuropatia.[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Sintomas cardíacos
O envolvimento cardíaco é mais comumente associado à amiloidose do tipo AL e à amiloidose relacionada à transtirretina (TTR) (hereditária e do tipo selvagem).[42]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis: diagnosis and management. Clin Lymphoma Myeloma. 2005 Nov;6(3):208-19.
http://www.ncbi.nlm.nih.gov/pubmed/16354326?tool=bestpractice.com
[43]Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med. 2006 Sep 25;166(17):1805-13.
http://archinte.jamanetwork.com/article.aspx?articleid=410996
http://www.ncbi.nlm.nih.gov/pubmed/17000935?tool=bestpractice.com
[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
[54]Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018 Jan;28(1):10-21.
http://www.ncbi.nlm.nih.gov/pubmed/28739313?tool=bestpractice.com
[55]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[65]Muchtar E, Gertz MA, Kumar SK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017 Apr 13;129(15):2111-2119.
https://www.doi.org/10.1182/blood-2016-11-751628
http://www.ncbi.nlm.nih.gov/pubmed/28126928?tool=bestpractice.com
A tontura pode ser um sintoma de amiloidose cardíaca (baixo débito cardíaco com fração de ejeção preservada). Os pacientes podem apresentar claudicação da mandíbula, da panturrilha e dos membros e, raramente, angina, em caso de comprometimento de arteríolas coronárias.
A fadiga e a dispneia ao esforço, causadas por um comprometimento cardíaco inicial, geralmente não são reconhecidas como sintomas de insuficiência cardíaca evidente e podem ser diagnosticadas erroneamente como funcionais ou relacionadas ao estresse.
Envolvimento renal
Mais comumente associado à amiloidose do tipo AL, amiloidose AA, amiloidose hereditária não relacionada à TTR (por exemplo, cadeia alfa do fibrinogênio A, apolipoproteína A) e amiloidose por fator quimiotático 2 de leucócitos (LECT2).[3]Buxbaum JN, Dispenzieri A, Eisenberg DS, et al. Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2022 Dec;29(4):213-9.
https://www.tandfonline.com/doi/full/10.1080/13506129.2022.2147636
http://www.ncbi.nlm.nih.gov/pubmed/36420821?tool=bestpractice.com
[16]Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun 7;356(23):2361-71.
https://www.nejm.org/doi/10.1056/NEJMoa070265
http://www.ncbi.nlm.nih.gov/pubmed/17554117?tool=bestpractice.com
[33]Rezk T, Gilbertson JA, Rowczenio D, et al. Diagnosis, pathogenesis and outcome in leucocyte chemotactic factor 2 (ALECT2) amyloidosis. Nephrol Dial Transplant. 2018 Feb 1;33(2):241-7.
https://academic.oup.com/ndt/article/33/2/241/2527565?login=false
http://www.ncbi.nlm.nih.gov/pubmed/29401357?tool=bestpractice.com
[40]Gertz MA, Leung N, Lacy MQ, et al. Clinical outcome of immunoglobulin light chain amyloidosis affecting the kidney. Nephrol Dial Transplant. 2009 Oct;24(10):3132-7.
https://www.doi.org/10.1093/ndt/gfp201
http://www.ncbi.nlm.nih.gov/pubmed/19403931?tool=bestpractice.com
[52]Cervantes CE, Atta MG. Kidney amyloidosis: updates on pathogenesis and therapeutic frontiers. Am J Nephrol. 2024 Jun 12:1-12.
https://karger.com/ajn/article/doi/10.1159/000539596/909119/Kidney-Amyloidosis-Updates-on-Pathogenesis-and
http://www.ncbi.nlm.nih.gov/pubmed/38865984?tool=bestpractice.com
[60]Papa R, Lachmann HJ. Secondary, AA, amyloidosis. Rheum Dis Clin North Am. 2018 Nov;44(4):585-603.
http://www.ncbi.nlm.nih.gov/pubmed/30274625?tool=bestpractice.com
Fadiga e tontura são sintomas da síndrome nefrótica (hipoalbuminemia e contração do volume intravascular).
Sintomas neurológicos
O envolvimento nervoso pode levar à neuropatia periférica e autonômica e é mais comumente associado à amiloidose do tipo AL e à amiloidose por TTR hereditária (ATTRv).[19]Vaxman I, Gertz M. When to suspect a diagnosis of amyloidosis. Acta Haematol. 2020;143(4):304-11.
https://www.doi.org/10.1159/000506617
http://www.ncbi.nlm.nih.gov/pubmed/32340017?tool=bestpractice.com
[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
[66]Conceição I, González-Duarte A, Obici L, Schmidt HH, Simoneau D, Ong ML, Amass L. "Red-flag" symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016 Mar;21(1):5-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4788142
http://www.ncbi.nlm.nih.gov/pubmed/26663427?tool=bestpractice.com
[67]Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011 Dec;10(12):1086-97.
http://www.ncbi.nlm.nih.gov/pubmed/22094129?tool=bestpractice.com
[68]Kaku M, Berk JL. Neuropathy associated with systemic amyloidosis. Semin Neurol. 2019 Oct;39(5):578-88.
http://www.ncbi.nlm.nih.gov/pubmed/31639841?tool=bestpractice.com
[69]Gonzalez-Duarte A, Valdés-Ferrer SI, Cantú-Brito C. Characteristics and natural history of autonomic involvement in hereditary ATTR amyloidosis: a systematic review. Clin Auton Res. 2019 Sep;29(suppl 1):1-9.
https://www.doi.org/10.1007/s10286-019-00630-y
http://www.ncbi.nlm.nih.gov/pubmed/31473866?tool=bestpractice.com
A neuropatia periférica leve pode ocorrer em pacientes com amiloidose por ATTRwt (até 20%).[56]Grogan M, Scott CG, Kyle RA, et al. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J Am Coll Cardiol. 2016 Sep 6;68(10):1014-20.
https://www.doi.org/10.1016/j.jacc.2016.06.033
http://www.ncbi.nlm.nih.gov/pubmed/27585505?tool=bestpractice.com
O envolvimento nervoso não é uma característica típica da amiloidose AA, amiloidose hereditária não associada à TTR ou amiloidose por LECT2.[3]Buxbaum JN, Dispenzieri A, Eisenberg DS, et al. Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2022 Dec;29(4):213-9.
https://www.tandfonline.com/doi/full/10.1080/13506129.2022.2147636
http://www.ncbi.nlm.nih.gov/pubmed/36420821?tool=bestpractice.com
Geralmente, a apresentação inicial da neuropatia periférica é a perda sensorial distal simétrica (ou seja, perda da percepção de temperatura e da dor, seguida de perda proprioceptiva).[51]Kapoor M, Rossor AM, Jaunmuktane Z, et al. Diagnosis of amyloid neuropathy. Pract Neurol. 2019 Jun;19(3):250-8.
http://www.ncbi.nlm.nih.gov/pubmed/30598431?tool=bestpractice.com
Os pacientes geralmente relatam disestesia e parestesia dos pés e pernas, que progridem para as mãos e braços ao longo do tempo.
Pacientes com neuropatia autonômica podem apresentar disfunção erétil, hipotensão ortostática, disfunção gastrointestinal ou disfunção urinária.[19]Vaxman I, Gertz M. When to suspect a diagnosis of amyloidosis. Acta Haematol. 2020;143(4):304-11.
https://www.doi.org/10.1159/000506617
http://www.ncbi.nlm.nih.gov/pubmed/32340017?tool=bestpractice.com
[69]Gonzalez-Duarte A, Valdés-Ferrer SI, Cantú-Brito C. Characteristics and natural history of autonomic involvement in hereditary ATTR amyloidosis: a systematic review. Clin Auton Res. 2019 Sep;29(suppl 1):1-9.
https://www.doi.org/10.1007/s10286-019-00630-y
http://www.ncbi.nlm.nih.gov/pubmed/31473866?tool=bestpractice.com
Anormalidades de sudorese e insuficiência da frequência cardíaca ao mudar a posição corporal são sinais de disfunção autonômica.
As neuropatias periférica e autonômica são importantes pistas diagnósticas para a amiloidose do tipo AL e para a amiloidose do tipo ATTRv.[51]Kapoor M, Rossor AM, Jaunmuktane Z, et al. Diagnosis of amyloid neuropathy. Pract Neurol. 2019 Jun;19(3):250-8.
http://www.ncbi.nlm.nih.gov/pubmed/30598431?tool=bestpractice.com
A prevalência da síndrome do túnel do carpo é maior em pacientes com amiloidose cardíaca por TTR (20.3% vs. 4.1% na população em geral), mas também é uma manifestação da amiloidose do tipo AL.[70]Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020 Mar;22(3):507-15.
https://onlinelibrary.wiley.com/doi/10.1002/ejhf.1742
http://www.ncbi.nlm.nih.gov/pubmed/31975495?tool=bestpractice.com
Deve-se pesquisar o sinal de Tinel (a percussão sobre o nervo do carpo no punho produz formigamento nos dedos polegar, indicador e médio) e realizar a manobra de Phalen (o ato de manter a superfície dorsal das mãos unidas em flexão forçada por cerca de 1 minuto resulta em formigamento nos dedos polegar, indicador e médio) para se testar se há comprometimento pela síndrome do túnel do carpo nos pacientes que se queixarem de parestesia nas mãos.
Sintomas gastrointestinais
A esteatorreia é típica de comprometimento intestinal. Pode haver incontinência fecal grave alternando com 3-4 dias de constipação.
Sintomas pseudo-obstrutivos (incluindo náuseas, vômitos, cólicas abdominais pós-prandiais) e gastroparesia podem estar presentes se houver envolvimento do trato gastrointestinal superior.
Sintomas musculoesqueléticos
Os distúrbios musculoesqueléticos (por exemplo, ruptura do tendão do bíceps, osteoartrite de quadril e joelho, dedo em gatilho e estenose da coluna vertebral) estão tipicamente associados à amiloidose por TTR e podem preceder as manifestações cardíacas ou neurológicas.[71]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[72]Nativi-Nicolau JN, Karam C, Khella S, et al. Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev. 2022 May;27(3):785-93.
https://link.springer.com/article/10.1007/s10741-021-10080-2
http://www.ncbi.nlm.nih.gov/pubmed/33609196?tool=bestpractice.com
Exame físico
Os achados físicos comuns incluem edema de membros inferiores e distensão venosa jugular elevada (devido à alta pressão de enchimento do lado direito).[73]Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995 Jan;32(1):45-59.
http://www.ncbi.nlm.nih.gov/pubmed/7878478?tool=bestpractice.com
[74]Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005 Sep 27;112(13):2047-60.
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.104.489187?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/16186440?tool=bestpractice.com
Muitos achados físicos da amiloidose são específicos e têm valor diagnóstico para a amiloidose, mas estão presentes em ≤15% dos pacientes.[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Púrpura amiloide: ocorre em aproximadamente 15% dos pacientes com amiloidose do tipo AL.[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Normalmente periorbital, mas pode ocorrer em qualquer lugar acima da linha mamilar.
Petéquias palpebrais: comuns, mas evidentes somente quando os olhos do paciente estão fechados.
Macroglossia: específica e diagnóstica de amiloidose do tipo AL.[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Ocorre em aproximadamente 10% dos pacientes, mas não é facilmente percebida, pois a apresentação mais comum são as endentações dentárias na parte de baixo da língua.
Aumento das glândulas salivares submandibulares: específico para amiloidose do tipo AL. Pode ser interpretado erroneamente como linfadenopatia. O comprometimento da glândula salivar resulta em uma síndrome seca. Esses pacientes muitas vezes são diagnosticados erroneamente como tendo síndrome de Sjögren.
Hepatomegalia palpável: >5 cm abaixo da margem costal direita, relatada com mais frequência na amiloidose do tipo AL (aproximadamente 10% dos pacientes).[16]Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007 Jun 7;356(23):2361-71.
https://www.nejm.org/doi/10.1056/NEJMoa070265
http://www.ncbi.nlm.nih.gov/pubmed/17554117?tool=bestpractice.com
[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
Geralmente, a esplenomegalia tem grau modesto. A hepatomegalia palpável é rara na amiloidose AA, mas alterações histopatológicas no fígado podem ser evidentes. O comprometimento hepático é raro em pacientes com amiloidose hereditária.
Ombros com aspecto de ombreira (sinal de "shoulder pad"): raros, em decorrência de infiltração periarticular com amiloide; a pseudo-hipertrofia é específica da amiloidose do tipo AL.
Fraqueza muscular difusa: pode ocorrer miopatia amiloide com hipertrofia muscular em decorrência de infiltração extracelular de amiloide no músculo, ou com atrofia muscular devida à oclusão vascular, que resulta em isquemia muscular e claudicação.
Hipotensão ortostática com síncope: pode ocorrer se houver neuropatia autonômica (por exemplo, na amiloidose do tipo AL ou amiloidose por ATTRv).
[Figure caption and citation for the preceding image starts]: Equimose periorbital bilateral (púrpura amiloide) em paciente com amiloidose do tipo ALWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends]. [Figure caption and citation for the preceding image starts]: Púrpura periorbital clássicaMorie A. Gertz, MD; cortesia da Mayo Clinic [Citation ends].
[Figure caption and citation for the preceding image starts]: Púrpura periorbital clássicaMorie A. Gertz, MD; cortesia da Mayo Clinic [Citation ends].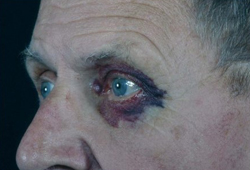 [Figure caption and citation for the preceding image starts]: Macroglossia em paciente com amiloidose do tipo ALWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends].
[Figure caption and citation for the preceding image starts]: Macroglossia em paciente com amiloidose do tipo ALWilliams MU, Murphy CE, Gore RS, et al. BMJ Case Rep 2018;11:e225923. doi:10.1136/bcr-2018- 225923 [Citation ends].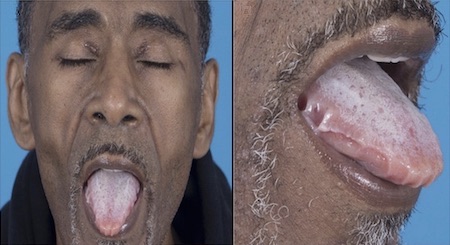
Principais exames diagnósticos
Os primeiros exames a serem solicitados em pacientes com suspeita clínica de amiloidose são a eletroforese de proteínas e imunofixação no soro e na urina (usando coleta de urina de 24 horas) e o ensaio da cadeia leve livre de imunoglobulina sérica.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
[75]Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009 Feb;23(2):215-24.
http://www.ncbi.nlm.nih.gov/pubmed/19020545?tool=bestpractice.com
Imunofixação positiva (presença de uma proteína monoclonal no soro ou urina) e/ou um ensaio anormal da cadeia leve livre de imunoglobulina sérica são relatados em 99% dos pacientes com amiloidose do tipo AL.[76]Katzmann JA, Abraham RS, Dispenzieri A, et al. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem. 2005 May;51(5):878-81.
https://www.doi.org/10.1373/clinchem.2004.046870
http://www.ncbi.nlm.nih.gov/pubmed/15774572?tool=bestpractice.com
O diagnóstico de amiloidose do tipo AL deve ser confirmado histologicamente (por exemplo, biópsia com tipagem amiloide) para evitar um diagnóstico incorreto, pois uma gamopatia monoclonal incidental pode estar presente em outros tipos de amiloidose (por exemplo, amiloidose hereditária, amiloidose por ATTRwt).[28]Comenzo RL, Zhou P, Fleisher M, et al. Seeking confidence in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood. 2006 May 1;107(9):3489-91.
http://www.bloodjournal.org/content/107/9/3489.full
http://www.ncbi.nlm.nih.gov/pubmed/16439680?tool=bestpractice.com
[29]Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002 Jun 6;346(23):1786-91.
http://www.nejm.org/doi/full/10.1056/NEJMoa013354#t=article
http://www.ncbi.nlm.nih.gov/pubmed/12050338?tool=bestpractice.com
[61]Phull P, Sanchorawala V, Connors LH, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018 Mar;25(1):62-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6157907
http://www.ncbi.nlm.nih.gov/pubmed/29424556?tool=bestpractice.com
[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
[77]Morris KL, Tate JR, Gill D, et al. Diagnostic and prognostic utility of the serum free light chain assay in patients with AL amyloidosis. Intern Med J. 2007 Jul;37(7):456-63.
http://www.ncbi.nlm.nih.gov/pubmed/17547724?tool=bestpractice.com
O diagnóstico de amiloidose do tipo AL é improvável se a imunofixação e o ensaio da cadeia leve livre de imunoglobulina sérica forem normais. Pacientes com suspeita clínica de amiloidose com imunofixação duvidosa ou normal e ensaio de cadeia leve livre de imunoglobulina sérica devem ser submetidos a uma avaliação cuidadosa e imediata (incluindo testes genéticos) para outros tipos de amiloidose (por exemplo, amiloidose AA, amiloidose por TTR, amiloidose localizada).
Estudos de biópsia
A confirmação histológica de depósitos amiloides no tecido é essencial para estabelecer o diagnóstico de amiloidose.
O aspirado e biópsia de medula óssea e aspirado de gordura subcutânea (por exemplo, coxim gorduroso abdominal) são recomendados em pacientes com suspeita de amiloidose (por exemplo, se uma proteína monoclonal estiver presente).[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Outros tecidos que podem ser submetidos à biópsia incluem lábio (glândula salivar menor) e reto.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Aspirado e biópsia de medula óssea também podem ser usados para identificar plasmócitos clonais e avaliar a presença de mieloma múltiplo coexistente. Consulte Mieloma múltiplo.
Se os estudos de biópsia de medula óssea e tecido forem negativos, a biópsia de um órgão envolvido (por exemplo, coração, fígado, rim, nervo) deve ser realizada conforme indicação clínica.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Biópsias de múltiplos tecidos ou órgãos são potencialmente perigosas e não são recomendadas.[78]Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015 Jan;168(2):207-18.
http://onlinelibrary.wiley.com/doi/10.1111/bjh.13156/full
http://www.ncbi.nlm.nih.gov/pubmed/25312307?tool=bestpractice.com
A biópsia da medula óssea combinada com aspiração de gordura subcutânea (por exemplo, coxim gorduroso abdominal) identificará depósitos amiloides na maioria (85%) dos pacientes com amiloidose.[79]Muchtar E, Dispenzieri A, Lacy MQ, et al. Overuse of organ biopsies in immunoglobulin light chain amyloidosis (AL): the consequence of failure of early recognition. Ann Med. 2017 Nov;49(7):545-551.
https://www.doi.org/10.1080/07853890.2017.1304649
http://www.ncbi.nlm.nih.gov/pubmed/28271734?tool=bestpractice.com
A birrefringência verde-maçã em um aspirado ou amostra de biópsia corada com vermelho Congo é necessária para o diagnóstico.[80]Gertz MA. The classification and typing of amyloid deposits. Am J Clin Pathol. 2004 Jun;121(6):787-9.
http://www.ncbi.nlm.nih.gov/pubmed/15198347?tool=bestpractice.com
A birrefringência verde-maçã após coloração com vermelho Congo confirma a presença de depósitos de amiloide, mas não diferencia entre diferentes tipos de amiloide.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
A tipagem amiloide e/ou estudos imuno-histoquímicos devem ser realizados para confirmar o tipo de amiloide.
Devem-se realizar estudos de hibridização in situ fluorescente (FISH) nos aspirados de medula óssea para identificar marcadores moleculares que possam orientar o prognóstico e o tratamento, por exemplo translocação t(11;14).[81]Gertz MA, Dispenzieri A, Muchtar E. Importance of FISH genetics in light chain amyloidosis. Oncotarget. 2017 Oct 10;8(47):81735-6.
https://pmc.ncbi.nlm.nih.gov/articles/PMC5669844
[82]Muchtar E, Dispenzieri A, Kumar SK, et al. Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia. 2017 Jul;31(7):1562-9.
http://www.ncbi.nlm.nih.gov/pubmed/27904139?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Vaso sanguíneo com coloração vermelho Congo em biópsia da medula óssea demonstrando birrefringência verde patognomônica de amiloidoseMorie A. Gertz, MD; cortesia da Mayo Clinic [Citation ends].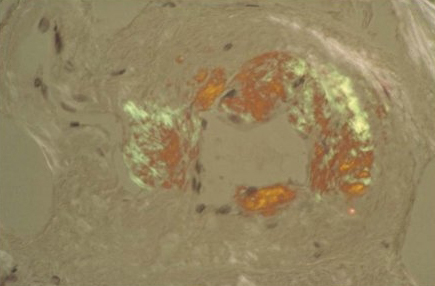
Tipagem do amiloide
Atualmente, a análise proteômica baseada na espectroscopia de massas é o padrão ouro para descobrir o tipo de amiloide. É o método mais direto de confirmação do tipo amiloide (por exemplo, cadeia leve, amiloide A sérica [associada à amiloidose AA], TTR).
A microscopia imunoeletrônica pode ser usada em amostras de biópsia renal, a fim de esclarecer a natureza fibrilar do amiloide, mas não faz parte da prática clínica de rotina para outros materiais de biópsia.[Figure caption and citation for the preceding image starts]: Eletromicrografia demonstrando fibras amiloides clássicasMorie A. Gertz, MD; cortesia da Mayo Clinic [Citation ends].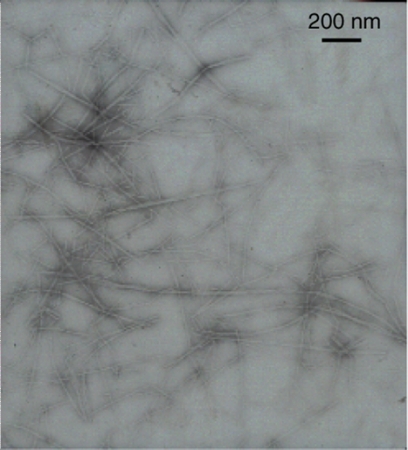
Estudos imuno-histoquímicos
Pode-se realizar uma coloração imuno-histoquímica dos depósitos de amiloide a fim de distinguir as diversas formas de amiloidose sistêmica. Geralmente, são utilizados antissoros para cadeias leves de imunoglobulina, amiloide A sérica e TTR, disponíveis comercialmente, mas podem apresentar ausência de especificidade e sensibilidade.
A imuno-histoquímica tem menor precisão diagnóstica do que a espectrometria de massa.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Teste genético
Testes genéticos podem ser usados para avaliar amiloidose hereditária (por exemplo, ATTRv, cadeia alfa do fibrinogênio A, apolipoproteína A, lisozima) e síndrome febril periódica hereditária associada à amiloidose AA (por exemplo, febre familiar do Mediterrâneo, TRAPS, CAPS [síndrome de Muckle-Wells], deficiência de mevalonato-quinase [síndrome de hiperimunoglobulinemia D]).[83]Gillmore JD, Reilly MM, Coats CJ, et al. Clinical and genetic evaluation of people with or at risk of hereditary ATTR amyloidosis: an expert opinion and consensus on best practice in Ireland and the UK. Adv Ther. 2022 Jun;39(6):2292-301.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9122857
http://www.ncbi.nlm.nih.gov/pubmed/35419651?tool=bestpractice.com
[84]Conceição I, Damy T, Romero M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019 Mar;26(1):3-9.
https://www.tandfonline.com/doi/full/10.1080/13506129.2018.1556156
http://www.ncbi.nlm.nih.gov/pubmed/30793974?tool=bestpractice.com
O uso de testes genéticos é importante para evitar um diagnóstico incorreto (por exemplo, amiloidose do tipo AL) em pacientes com amiloidose hereditária.[30]Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020 Sep 23;21(1):198.
https://www.doi.org/10.1186/s12875-020-01252-4
http://www.ncbi.nlm.nih.gov/pubmed/32967612?tool=bestpractice.com
[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Exames auxiliares
Os pacientes devem fazer os seguintes exames para orientar o diagnóstico e o prognóstico e para avaliar o envolvimento de órgãos:[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Hemograma completo com diferencial
Esfregaço de sangue periférico, imunoglobulinas quantitativas séricas e eletroforese de proteínas séricas para avaliar distúrbios plasmocíticos
Fragmento N-terminal do peptídeo natriurético tipo B (NT-proPNB; peptídeo natriurético do tipo B [PNB], se o NT-proPNB não estiver disponível), troponina T sérica (troponina I, se troponina T não estiver disponível) e perfil lipídico, para avaliar o envolvimento cardíaco e para prognóstico
Proteína total na urina e eletroforese de proteínas urinárias (da amostra de urina de 24 horas)
Perfil metabólico abrangente (incluindo ureia sérica, creatinina sérica, eletrólitos, albumina sérica, cálcio sérico, ácido úrico sérico, lactato desidrogenase sérica [LDH], beta-2 microglobulina; testes da função hepática [TFHs]), para avaliar o envolvimento renal e hepático
Estudos de coagulação (incluindo tempo de protrombina [TP], tempo de tromboplastina parcial [TTP], fator X), para avaliar anormalidades de coagulação relacionadas a amiloide
Avaliação dos sinais vitais ortostáticos e eletromiografia (se houver neuropatia periférica clinicamente significativa)/estudos de condução nervosa, para avaliar o envolvimento dos nervos
Níveis de hormônio estimulante da tireoide e cortisol, para avaliar o envolvimento endócrino
Testes da função pulmonar, para avaliar o envolvimento pulmonar
Avaliação cardíaca
O envolvimento cardíaco ocorre comumente na amiloidose do tipo AL, ATTRv e ATTRwt.[42]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis: diagnosis and management. Clin Lymphoma Myeloma. 2005 Nov;6(3):208-19.
http://www.ncbi.nlm.nih.gov/pubmed/16354326?tool=bestpractice.com
[43]Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med. 2006 Sep 25;166(17):1805-13.
http://archinte.jamanetwork.com/article.aspx?articleid=410996
http://www.ncbi.nlm.nih.gov/pubmed/17000935?tool=bestpractice.com
[49]Gertz MA, Lacy MQ, Dispenzieri A, et al. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27.
http://www.ncbi.nlm.nih.gov/pubmed/16026746?tool=bestpractice.com
[54]Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018 Jan;28(1):10-21.
http://www.ncbi.nlm.nih.gov/pubmed/28739313?tool=bestpractice.com
[55]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[65]Muchtar E, Gertz MA, Kumar SK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017 Apr 13;129(15):2111-2119.
https://www.doi.org/10.1182/blood-2016-11-751628
http://www.ncbi.nlm.nih.gov/pubmed/28126928?tool=bestpractice.com
O retardo no diagnóstico de um comprometimento cardíaco está associado a desfechos desfavoráveis.[85]Tuzovic M, Yang EH, Baas AS, et al. Cardiac amyloidosis: diagnosis and treatment strategies. Curr Oncol Rep. 2017 Jul;19(7):46.
http://www.ncbi.nlm.nih.gov/pubmed/28528458?tool=bestpractice.com
Um ECG e ecocardiografia (com Doppler tecidual e strain longitudinal global) devem ser realizados em todos os pacientes (sintomáticos ou assintomáticos) com amiloidose cardíaca suspeita ou confirmada.[55]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[71]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[86]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
O aumento da espessura da parede do ventrículo esquerdo (VE), o padrão de strain longitudinal típico do VE e a redução da velocidade do Doppler tecidual são altamente sugestivos de amiloidose.[55]Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020 Jul 7;142(1):e7-22.
https://www.doi.org/10.1161/CIR.0000000000000792
http://www.ncbi.nlm.nih.gov/pubmed/32476490?tool=bestpractice.com
[86]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
A RNM cardíaca pode ser útil se a ecocardiografia for sugestiva ou indeterminada. No entanto, ela não é diagnóstica e não consegue distinguir entre amiloidose cardíaca do tipo AL e TTR.[71]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[86]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
Os parâmetros da RNM cardíaca devem ser combinados com achados eletrocardiográficos, clínicos, biomarcadores e outros exames de imagem para maximizar a acurácia diagnóstica.[86]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
A cintilografia cardíaca com marcadores ósseos marcados com tecnécio (99mTc-PYP ou 99mTc-DPD) deve ser realizada se houver suspeita de amiloidose cardíaca por TTR.[30]Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020 Sep 23;21(1):198.
https://www.doi.org/10.1186/s12875-020-01252-4
http://www.ncbi.nlm.nih.gov/pubmed/32967612?tool=bestpractice.com
A cintilografia é sensível para a detecção de amiloidose cardíaca do tipo TTR e permite o diagnóstico clínico não invasivo.[71]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
[86]Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000029.
https://www.ahajournals.org/doi/full/10.1161/HCI.0000000000000029?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/34196223?tool=bestpractice.com
[87]Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016 Jun 14;133(24):2404-12.
https://www.doi.org/10.1161/CIRCULATIONAHA.116.021612
http://www.ncbi.nlm.nih.gov/pubmed/27143678?tool=bestpractice.com
[88]Castano A, Haq M, Narotsky DL, et al. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol. 2016 Nov 1;1(8):880-9.
https://www.doi.org/10.1001/jamacardio.2016.2839
http://www.ncbi.nlm.nih.gov/pubmed/27557400?tool=bestpractice.com
Entretanto, ela carece de especificidade, portanto, falsos-positivos podem ser observados. Um resultado negativo de proteína monoclonal juntamente com captação cardíaca na cintilografia (grau 2 ou 3) confirma o diagnóstico de amiloidose por TTR (sem necessidade de biópsia).[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
[71]Writing Committee, Kittleson MM, Ruberg FL, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology solution set oversight committee. J Am Coll Cardiol. 2023 Mar 21;81(11):1076-1126.
https://www.sciencedirect.com/science/article/pii/S073510972207423X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/36697326?tool=bestpractice.com
Se a captação da cintilografia cardíaca for 1 ou 0, será necessária biópsia (cardíaca ou não cardíaca, dependendo da apresentação).[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Em homens mais velhos com uma ecocardiografia condizente com amiloidose cardíaca, uma cintilografia cardíaca que mostre captação de 99mTc-PYP ou 99mTc-DPD no tecido miocárdico aumenta a suspeita de amiloidose por ATTRwt.
A investigação cardíaca em busca de alguma doença arterial coronariana é invariavelmente normal.[89]Neben-Wittich MA, Wittich CM, Mueller PS, et al. Obstructive intramural coronary amyloidosis and myocardial ischemia are common in primary amyloidosis. Am J Med. 2005 Nov;118(11):1287.
http://www.ncbi.nlm.nih.gov/pubmed/16271914?tool=bestpractice.com
Exames de imagem
A tomografia computadorizada (TC) de corpo inteiro de baixa dose ou a tomografia por emissão de pósitrons com 18F-fluordesoxiglucose (FDG-PET)/TC podem ser usadas para detectar lesões ósseas osteolíticas se uma proteína monoclonal for detectada. Uma radiografia de esqueleto pode ser usada se essas modalidades avançadas de imagem não estiverem disponíveis, mas a sensibilidade é significativamente menor.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Uma TC de tórax pode ser usada para avaliar o envolvimento pulmonar, se clinicamente indicado.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
A cintilografia de esvaziamento gástrico (se houver gastroparesia) e a ultrassonografia ou TC abdominal (conforme indicação clínica) podem ser usadas para avaliar o envolvimento hepático e gastrointestinal.[62]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].
https://www.nccn.org/guidelines/category_1
Endoscopias alta e baixa podem ser realizadas se os sintomas sugerirem envolvimento gastrointestinal.
A cintilografia com amiloide sérico P marcado com 123I (SAP) pode ser usada para avaliar a extensão do envolvimento e disfunção do órgão no diagnóstico e durante o acompanhamento. A cintilografia com SAP é uma prática padrão no Reino Unido e na Holanda, mas o uso em outros países pode variar.[78]Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015 Jan;168(2):207-18.
http://onlinelibrary.wiley.com/doi/10.1111/bjh.13156/full
http://www.ncbi.nlm.nih.gov/pubmed/25312307?tool=bestpractice.com
[90]Hawkins PN, Lavender JP, Pepys MB. Evaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P component. N Engl J Med. 1990 Aug 23;323(8):508-13.
https://www.doi.org/10.1056/NEJM199008233230803
http://www.ncbi.nlm.nih.gov/pubmed/2377176?tool=bestpractice.com
Prognóstico
Os biomarcadores prognósticos para amiloidose incluem: troponina T sérica (troponina I, se a troponina T não estiver disponível); NT-proPNB (PNB, se NT-proPNB não estiver disponível); e a diferença entre cadeias leves livres no soro envolvidas e não envolvidas (dFLC).
O nível de troponina sérica é um marcador sensível para lesão miocárdica na amiloidose; NT-proPNB é um marcador sensível para estiramento miocárdico e insuficiência cardíaca congestiva.[91]Dispenzieri A, Kyle RA, Gertz MA, et al. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet. 2003 May 24;361(9371):1787-9.
http://www.ncbi.nlm.nih.gov/pubmed/12781539?tool=bestpractice.com
[92]Palladini G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003 May 20;107(19):2440-5.
https://www.ahajournals.org/doi/abs/10.1161/01.CIR.0000068314.02595.B2
http://www.ncbi.nlm.nih.gov/pubmed/12719281?tool=bestpractice.com
Troponina, NT-proPNB e dFLC são usados nos critérios de estadiamento de Mayo para amiloidose do tipo AL.[93]Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004 Sep 15;22(18):3751-7.
http://www.ncbi.nlm.nih.gov/pubmed/15365071?tool=bestpractice.com
[94]Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012 Mar 20;30(9):989-95.
http://www.ncbi.nlm.nih.gov/pubmed/22331953?tool=bestpractice.com
[95]Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013 Apr 25;121(17):3420-7.
https://www.doi.org/10.1182/blood-2012-12-473066
http://www.ncbi.nlm.nih.gov/pubmed/23479568?tool=bestpractice.com
Consulte Critérios.
A beta-2-microglobulina é preditiva de sobrevida em pacientes com amiloidose do tipo AL.[96]Zerbini CA, Anderson JJ, Kane KA, et al. Beta 2 microglobulin serum levels and prediction of survival in AL amyloidosis. Amyloid. 2002 Dec;9(4):242-6.
http://www.ncbi.nlm.nih.gov/pubmed/12557752?tool=bestpractice.com
[97]Al Saleh AS, Sidiqi MH, Muchtar E, et al. Prognostic role of beta-2 microglobulin in patients with light chain amyloidosis treated with autologous stem cell transplantation. Biol Blood Marrow Transplant. 2020 Aug;26(8):1402-1405.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7371505
http://www.ncbi.nlm.nih.gov/pubmed/32422250?tool=bestpractice.com