Etiologia
A doença de Graves é uma doença autoimune. A etiologia da superprodução do hormônio tireoidiano é a estimulação da tireoide pelos anticorpos antirreceptor de hormônio estimulante da tireoide (TSH).[21][22] Embora outros anticorpos antitireoidianos ocorram nas pessoas com doença de Graves (sejam eles anticorpos antitireoglobulina [Tg], antitireoperoxidase [TPO] e contra o transportador de iodeto de sódio), eles não desempenham nenhum papel importante no desenvolvimento do hipertireoidismo.[23] A causa fundamental da autoimunidade não é clara, mas as evidências dão suporte a um componente genético significativo.[3]
A doença de Graves é causada por uma combinação de fatores genéticos e ambientais em um indivíduo imunologicamente suscetível.[3] A taxa de concordância da doença para gêmeos monozigóticos é maior do que a relatada para gêmeos dizigóticos.[24][25] Estudos com gêmeos e famílias demonstram que a doença de Graves não é causada por um único defeito gênico, mas por padrões de herança complexos. Várias regiões gênicas foram associadas ao hipertireoidismo de Graves, incluindo o alelo do antígeno leucocitário humano (HLA)-DRB1*16:02.[26][27]
O sexo feminino é um fator de risco. O consumo moderado de bebidas alcoólicas é associado a um risco reduzido para a doença de Graves, enquanto o tabagismo aumenta esse risco.[28][29][30] A doença de Graves é um efeito adverso comum do alentuzumabe, um medicamento usado para tratar a esclerose múltipla.[31]
Fisiopatologia
A doença de Graves é parte do espectro de doenças tireoidianas autoimunes, que também inclui a tireoidite de Hashimoto (infiltração linfocítica e destruição de tecido tireoidiano por anticorpos secundários contra tireoperoxidase, tireoglobulina e outros antígenos tireoidianos), a tireoidite indolor (destruição subaguda dos folículos tireoidianos, com liberação do hormônio pré-formado) e hipotireoidismo devido a anticorpos bloqueadores antirreceptor de hormônio estimulante da tireoide (TSH).[2] Muitos pacientes apresentam autoimunidade de sobreposição; assim, a maioria dos pacientes com doença de Graves também apresenta nível elevado de anticorpos antitireoperoxidase (TPO).[2]
As células foliculares da tireoide expressam moléculas do antígeno leucocitário humano (HLA) classe II em resposta à gamainterferona produzida pelas células T infiltrantes. Isso permite a apresentação de receptores de TSH às células T ativadas e o início da cascata autoimune. Os autoanticorpos do receptor de TSH causam hiperprodução do hormônio tireoidiano, bem como hipertrofia tireoidiana e hiperplasia das células foliculares da tireoide, resultando em manifestações clínicas de hipertireoidismo e bócio difuso.[3]
O autoanticorpo antirreceptor de TSH no soro pode ser medido por bioensaio, utilizando a geração de AMP cíclico em uma linhagem celular da tireoide (comumente chamada de imunoglobulina estimulante da tireoide [TSI] nos EUA), ou por meio de imunoensaio (chamado de imunoglobulina inibidora da ligação à tireotrofina [TBII] ou, genericamente, de anticorpo antirreceptor de tireotrofina [TRAb]). A atividade se correlaciona com a gravidade do processo autoimune.[2] A presença de anticorpos bloqueadores e o dano coexistente ao tecido tireoidiano como um componente da tireoidite autoimune podem modificar o estado hipertireoidiano.[3] Alguns pacientes têm orbitopatia de Graves, dermopatia e/ou acropaquia e são eutireóideos ou, até mesmo, hipotireóideos, embora apresentem níveis muito elevados de anticorpos antirreceptores de TSH.[10][32]
Manifestações extratireoidianas
A patogênese das manifestações extratireoidianas (por exemplo, orbitopatia, dermatopatia e acropaquia) é menos clara. Os receptores de TSH são encontrados em uma variedade de sítios extratireoidianos, em particular nos tecidos retro-orbitário e dérmico.[3] A estimulação de fibroblastos por autoanticorpos antirreceptor de TSH e citocinas produz glicosaminoglicanos que desencadeiam a expansão da gordura orbital e do tecido.[3] O sinergismo do fator de crescimento semelhante à insulina 1 (IGF-1) e os autoanticorpos antirreceptor de TSH estão implicados na patogênese da orbitopatia (e dermopatia).[3]
As manifestações clínicas da orbitopatia (como proptose, quemose e oftalmoplegia restritiva) são as consequências do aumento dos tecidos moles orbitais e do volume do músculo extraocular.[33] Os tecidos orbitais, incluindo músculos, são infiltrados pelas células inflamatórias, incluindo linfócitos, mastócitos e macrófagos, podendo causar retorno venoso insuficiente e, em casos extremos, pressão sobre o nervo óptico e exposição e ulceração da córnea.[3][33][34][Figure caption and citation for the preceding image starts]: Retração da pálpebra, proptose leve e quemose leveCedida pelo Dr. Vahab Fatourechi [Citation ends].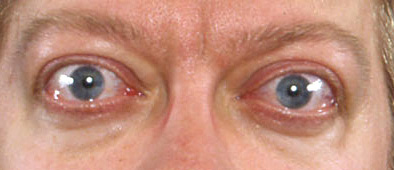 [Figure caption and citation for the preceding image starts]: Orbitopatia e elefantíaseCedida pelo Dr. Vahab Fatourechi [Citation ends].
[Figure caption and citation for the preceding image starts]: Orbitopatia e elefantíaseCedida pelo Dr. Vahab Fatourechi [Citation ends].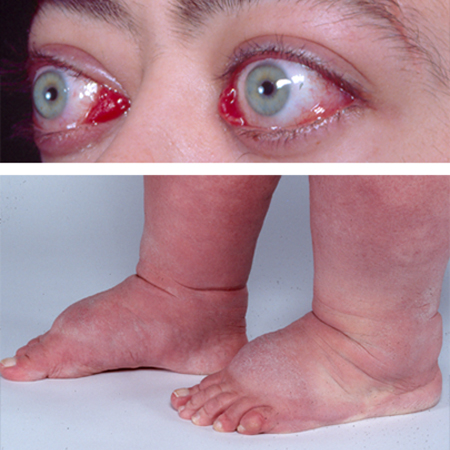
O uso deste conteúdo está sujeito ao nosso aviso legal