Etiologia
A etiologia da maioria das formas de aldosteronismo primário (AP) é desconhecida. A ocorrência de AP em famílias tem relação com a genética pelo menos em algumas formas dessa condição.[10][41][43] Uma variedade principalmente hereditária e remediável com glicocorticoide de AP é o hiperaldosteronismo familiar tipo I (HF-I). Descrito pela primeira vez em 1966, o HF-I é causado por um gene híbrido, que é composto por sequências do gene da 11-beta-hidroxilase (CYP11B1) em sua extremidade 5’ e por sequências do gene da aldosterona sintase (CYP11B2) em sua extremidade 3’.[44][45][Figure caption and citation for the preceding image starts]: Gene híbrido CYP11B1/CYP11B2 responsável pela produção em excesso de aldosterona regulada por hormônio adrenocorticotrófico (ACTH) em hiperaldosteronismo familiar tipo I (HF-I)Do acervo pessoal do Dr. Michael Stowasser; usado com permissão [Citation ends].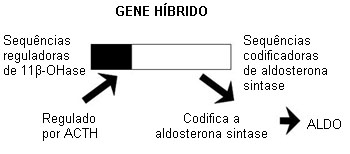
Em 1991, foi descrita uma segunda variedade familiar de AP, o hiperaldosteronismo familiar tipo II (HF-II), que não era remediável com glicocorticoides nem estava associado ao gene híbrido.[12][46] Embora o termo "HF-II" tenha sido originalmente usado para descrever formas familiares de AP diferentes do HF-I, este termo foi atribuído novamente ao AP familiar causado por mutação das linhas germinativas de CLCN2 após ter se revelado que a maior das famílias originalmente descritas (e vários outros probandos com AP) apresenta uma mutação nesse gene.[17] As características clínicas do HF-II incluem hipertensão de início precoce (<20 anos de idade), uma forma relativamente leve de AP em que a hipertensão é, em geral, prontamente controlada com medicamentos que bloqueiam a ação da aldosterona, anormalidades morfológicas mínimas ou nenhumas da adrenal em estudos de imagem, e falta de responsividade da aldosterona à postura ereta.
Uma família com PA precoce e grave, níveis acentuadamente elevados de "esteroides híbridos" (18-hidroxi- e 18-oxocortisol) e hiperplasia acentuada da zona fasciculada, relatada pela primeira vez em 2008 e designada como tendo hiperaldosteronismo familiar do tipo III (HF-III ), foi relatada como tendo uma mutação das linhas germinativas de KCNJ5 (codifica um canal de potássio).[14][15][47] As mutações de KCNJ5 nas linhagens germinativas parecem ser uma causa rara de AP bilateral e estão associadas a um início precoce (<18 anos de idade), mas com gravidade variável (de AP leve a bem intenso) que depende do tipo de mutação herdado.[16]
Mutações no CACNA 1A (que codifica um canal de cálcio dependente de voltagem) na linhagem germinativa também foram identificadas em um pequeno número de indivíduos com início de AP antes dos 10 anos de idade.[48] Em alguns desses indivíduos, a mutação foi herdada de um dos pais, dando origem ao termo "hiperaldosteronismo familiar do tipo IV" para descrever o AP decorrente de mutações de CACNA1H na linha germinativa. Mutações da linha germinativa em CACNA1D (também codifica um canal de cálcio dependente de voltagem) também foram encontradas em dois indivíduos com AP de início precoce (mas ainda sem parentes afetados conhecidos) que sofriam de convulsões e outros distúrbios neurológicos.[49]
Foi relatado que famílias com AP de etiologia genética incerta são pelo menos 5 vezes mais comuns do que o HF-I.[23] O adenoma produtor de aldosterona e a hiperplasia adrenal bilateral (HAB) são representados, normalmente dentro da mesma família.[13][22] Esses pacientes são clínica, bioquímica e morfologicamente indistinguíveis daqueles com AP aparentemente esporádico, e mutações responsáveis podem, portanto, estar subjacentes ao desenvolvimento de AP em outros pacientes sem história familiar dessa condição.[4][13][22][23]
Mutações somáticas em KCNJ5 foram identificadas em 8 de 22 adenomas produtores de aldosterona grandes e aparentemente esporádicos.[47] Outros grupos relataram a presença de mutações somáticas de KCNJ5 em 30% a 40% dos adenomas produtores de aldosterona removidos de pacientes brancos e em >60% dos removidos de pacientes do Japão e da China.[50][51][52] Desde então, mutações somáticas têm sido identificadas em ATP1A1 (codifica a subunidade α de Na+/K+ ATPase), ATP2B3 (um canal de cálcio Ca2+ ATPase) e CACNA1D (codifica um canal de cálcio dependente de voltagem) em proporções muito menores (5%, 2% e 11%, respectivamente) de adenomas produtores de aldosterona (APAs).[49][53][54]
Estudos que envolvem técnicas de imuno-histoquímica revelaram a presença de focos anormais de células de expressão de CYP11B2, denominadas aglomerados de células produtoras de aldosterona (APCCs), dentro dos córtices adrenais, que se tornam mais numerosos com o avançar da idade.[55] Simultaneamente, a zona glomerulosa, normalmente contínua na infância e no início da idade adulta, é cada vez mais atingida e relativamente suprimida em termos da expressão de CYP11B2. Estas observações respaldam um possível mecanismo fisiopatológico (formação de APCC) subjacente ao aparente desenvolvimento gradual da produção autônoma de aldosterona com o envelhecimento (como evidenciado pelo aumento de ARR) e que pode predispor ao desenvolvimento de AP evidente, devido à hiperplasia adrenal bilateral (HAB) ou ao adenoma produtor de aldosterona (APA) (ou ambos).
Fisiopatologia
Em todas as formas de aldosteronismo primário (AP), a produção de aldosterona excede as necessidades do corpo e é relativamente autônoma em relação a seu regulador crônico normal, o sistema renina-angiotensina II.[56] Isso resulta em excesso de reabsorção do sódio através dos canais de sódio epiteliais sensíveis à amilorida dentro dos néfrons distais, levando à hipertensão e à supressão da renina-angiotensina II. A perda urinária de íons de potássio e de hidrogênio, trocados por sódio no néfron distal, pode resultar em hipocalemia e alcalose metabólica caso seja grave e prolongada o suficiente. As causas exatas da produção autônoma e em excesso de aldosterona em adenoma produtor de aldosterona e hiperplasia adrenal bilateral são desconhecidas, mas fatores genéticos relacionados à regulação do crescimento celular cortical adrenal e/ou biossíntese de esteroides provavelmente estão envolvidos. Focos anormais de células de expressão de CYP11B2, denominadas APCCs, foram detectados nos córtices adrenais e tornam-se mais numerosos com o avançar da idade.[55] Simultaneamente, a renina torna-se progressivamente suprimida, o que não ocorre com a aldosterona, mas torna-se menos sensível ao sal, o que sugere que a produção de aldosterona por estes APCCs pode ser constitutiva e não reativa à renina. A significância clínica destes APCCs é incerta e inclui as possibilidades de que eles representem uma base patológica para HAB, precursores da APAs ou uma nova forma AP.[57]
Em hiperaldosteronismo familiar tipo I (HF-I), o gene híbrido causador codifica uma enzima híbrida de estrutura exclusiva que sintetiza a aldosterona, mas, diferente do CYP11B2, é regulado pelo hormônio adrenocorticotrópico (ACTH) e não por angiotensina II.[45] Desse modo, nos casos de HF-I, a produção de aldosterona é regulada por ACTH, não por angiotensina II, e pode ser suprimida e controlada com a administração de pequenas doses de glicocorticoides, como a dexametasona.[44]
As mutações em KCNJ5 (que codifica um canal de potássio de retificação interna) causam a redução da seletividade do canal de potássio/sódio e do influxo de sódio, predispondo à despolarização da membrana celular, ao aumento do influxo de cálcio, ao aumento da expressão dos genes que promovem a síntese de aldosterona e ao aumento da produção de aldosterona pelas células adrenocorticais.[16][47] Mutações em ATP1A1, ATP2B3, CACNA1H e CACNA1D também parecem compartilhar aumento no influxo de cálcio como mecanismo fisiopatológico comum para o aumento da produção autônoma de aldosterona.[49][53][54] Mutações em CLCN2 predispõem à despolarização da membrana celular e possivelmente também ao aumento do influxo de cálcio.[17] O modo como esses efeitos provocam a proliferação das células adrenais e o desenvolvimento do tumor continua sendo incerto.
Embora a morbidade no AP resulte principalmente da hipertensão, as evidências experimentais e clínicas sugerem que o excesso de aldosterona pode trazer sequelas cardiovasculares adversas (inclusive remodelagem e fibrose), independentemente dos efeitos hipertensivos.[58][59] Em estudos animais, o excesso de aldosterona e a alta ingestão de sal parecem ser necessários para a indução de fibrose cardíaca,[58] tendo a vasculite coronariana sido observada como manifestação precoce.[60] Esses efeitos puderam ser evitados por meio da administração de antagonistas do receptor de mineralocorticoide.[58][60] As doses de aldosterona usadas em estudos experimentais têm sido bem altas e, assim, os resultados desses estudos podem ter aplicação limitada à situações clínicas. No entanto, vários grupos têm demonstrado de modo convincente anormalidades na morfologia ou função cardiovascular em pacientes com AP que parecem ser desproporcionais à elevação da pressão arterial (PA).[59][61][62][63][64] Elas incluem:
Aumento do índice de massa ventricular esquerda e redução da função diastólica; ambos tiveram uma melhora significativa após o tratamento específico de AP[59][61]
Redução da perfusão miocárdica em repouso e durante exercício[62][63]
Aumento da dispersão reflexiva miocárdica (um marcador ecocardiográfico de fibrose miocárdica)[64]
Aumento da proteinúria (como evidência de dano glomerular renal)[65]
Maior incidência de eventos cardiovasculares, que foi revertida após tratamento cirúrgico ou clínico específico.[66][67]
Evidências de remodelagem ventricular esquerda também foram registradas em pessoas com HF-I geneticamente comprovado que tinham evidências bioquímicas de excesso de aldosterona, mas ainda não tinham desenvolvido hipertensão.[68]
Classificação
Classificação patológica[3][4][5]
Adenoma produtor de aldosterona (APA): tumor adrenocortical benigno com pelo menos 10 mm de diâmetro que produz aldosterona de forma autônoma.[Figure caption and citation for the preceding image starts]: Adenoma produtor de aldosteronaDo acervo pessoal do Dr. Michael Stowasser; usado com permissão [Citation ends].
 Os APAs podem ser subdivididos em não responsivos à angiotensina (como no clássico tumor de Conn) ou responsivos à angiotensina; a responsividade é definida como aumento de pelo menos 50% na aldosterona plasmática com relação ao valor basal durante 2 ou 3 horas de posição ereta após ficar em decúbito à noite ou durante uma infusão de angiotensina II.
Os APAs podem ser subdivididos em não responsivos à angiotensina (como no clássico tumor de Conn) ou responsivos à angiotensina; a responsividade é definida como aumento de pelo menos 50% na aldosterona plasmática com relação ao valor basal durante 2 ou 3 horas de posição ereta após ficar em decúbito à noite ou durante uma infusão de angiotensina II.Nódulo produtor de aldosterona (NPA): lesão adrenocortical benigna com menos de 10 mm de diâmetro que produz aldosterona de forma autônoma.
Carcinoma adrenocortical produtor de aldosterona (CACPA): um tumor adrenocortical maligno que produz aldosterona autonomamente.
Outras formas unilaterais: uma glândula adrenal que produz aldosterona de forma excessiva e autônoma, mas sem tumor distinto identificado no exame anatomopatológico. Em vez disso, a adrenal contém um ou vários micronódulos produtores de aldosterona (MPA ou MMPA) que apresentam coloração positiva para CYP11B2 por imuno-histoquímica, mas não são distinguíveis do córtex circundante pela coloração com hematoxilina-eosina. Mais raramente, a hiperplasia difusa produtora de aldosterona (HDPA) mostra uma ampla região contínua de células da zona glomerulosa hiperplásicas e positivas para CYP11B2.
Formas bilaterais: ambas as glândulas adrenais afetadas por hiperplasia difusa e/ou nodular e produtoras de aldosterona de forma autônoma e em excesso; inclui as formas não remediável com glicocorticoide (idiopática) e remediável com glicocorticoide. Raramente, a hiperplasia bilateral é macronodular com secreção autônoma com mais frequência de cortisol que de aldosterona.
Classificação funcional (voltada para o tratamento)[6][7][8][9][10]
Aldosteronismo primário (AP) unilateral:
Inclui APA, NPA, CACPA e hiperplasia adrenal unilateral (ou primária).
AP bilateral:
Não remediável com glicocorticoide: inclui hiperplasia adrenal bilateral (idiopática), que raramente é macronodular (hiperaldosteronismo não familiar, familiar tipo II e familiar tipo III), e APA bilateral.
Remediável com glicocorticoide (hiperaldosteronismo familiar tipo I).
Classificação familiar[4][10][11][12][13][14][15][16][17]
Hiperaldosteronismo familiar tipo I (HF-I; remediável com glicocorticoide; associado ao gene híbrido).
Hiperaldosteronismo familiar do tipo II (HF-II; não remediável com glicocorticoide; associado a mutações de CLCN2 na linha germinativa).
Hiperaldosteronismo familiar tipo III (HF-III; não remediável com glicocorticoide; associado a mutações de KCNJ5 na linha germinativa).
Hiperaldosteronismo familiar tipo IV (HF-IV; não remediável com glicocorticoide; associado a mutações de CACNA1H na linha germinativa).
AP aparentemente não familiar (sem parentes afetados conhecidos).
O uso deste conteúdo está sujeito ao nosso aviso legal