Abordaje
El abordaje de un neonato con genitales atípicos implica a un equipo multidisciplinar de especialistas pediátricos.[18] Suele incluir un endocrinólogo, un neonatólogo, un cirujano urológico, un psicólogo clínico y una enfermera especializada. El equipo multidisciplinar principal debe tener vínculos con un equipo más amplio, que incluya especialistas en genética, bioquímica, especialidades médicas de adultos relevantes, como ginecología y endocrinología, servicios sociales y, si es posible, un foro de ética clínica.[8] Es fundamental que exista comunicación continua con la familia, así como comunicación con el médico de atención primaria.[8][19] Los siguientes conceptos deben guiar la evaluación diagnóstica de los neonatos con genitales atípicos:[1][8][20]
Asignación del sexo. No se asigna un sexo hasta haber completado la evaluación. Se debe llamar al niño "bebé", no niño o niña. Se debe recomendar a la familia posponer la selección del nombre del bebé hasta que se le haya asignado el sexo. La asignación del sexo se puede realizar incluso si no hay un diagnóstico genético o endocrino subyacente claro.[21]
Tras la evaluación inicial y las pruebas diagnósticas, el bebé debe ser atendido en un centro con experiencia multidisciplinaria en la evaluación de neonatos con genitales atípicos. En algunas situaciones, la atención en un hospital local puede ser adecuada con una consulta temprana con un equipo multidisciplinario especializado en un centro regional.
Lo más adecuado es que las conversaciones con la familia sean dirigidas por un profesional del equipo, por lo general, el endocrinólogo. Es útil obtener el consentimiento de los padres para tratar sobre los resultados iniciales de pruebas diagnósticas en conjunto, en lugar de que les informen cuando se obtiene cada resultado, ya que los padres pueden poner un énfasis indebido en el resultado de un cariotipo o en el nivel de testosterona y no apreciar el grado en que estos deben ser interpretados a la luz de los resultados de otras pruebas diagnósticas.[21]
Se debe respetar y comprender las necesidades, el origen, la cultura y las expectativas de los padres. Se debe asegurar a los padres que los niños con un trastorno del desarrollo sexual (TDS) pueden llevar una vida exitosa y funcionar bien en la sociedad. Las decisiones relacionadas con la asignación del sexo las realiza el equipo multidisciplinar del TDS con los padres. Se acuerda un plan de manejo del alta. Es también importante informar a los padres sobre el desarrollo sexual esperado, ya que eso contribuye a anticipar problemas con prontitud.[21]
Antecedentes
Los antecedentes familiares pueden identificar a los miembros de la familia con problemas parecidos, lo cual indica una herencia autosómica recesiva o ligada al cromosoma X. Esto incluye un estudio específico de los antecedentes de consanguinidad, cirugía genital, infertilidad, virilización materna durante el embarazo (p. ej., cambio de voz o hirsutismo), virilización de una mujer en la pubertad (deficiencia de 5 alfa reductasa), o de otros niños nacidos con genitales atípicos. Una muerte neonatal inexplicable en un miembro de la familia puede indicar una TDS no diagnosticada asociada con insuficiencia suprarrenal.
La mayoría de las causas genéticas atípicas de genitales atípicos se deben a enfermedades autosómicas recesivas, como la hiperplasia suprarrenal congénita (HSC), la deficiencia de 5 alfa reductasa y otros defectos en la biosíntesis de la testosterona. Una herencia recesiva ligada al cromosoma X podría sugerir insensibilidad androgéna (el gen del receptor de andrógenos se encuentra en el cromosoma X). La historia prenatal debe incluir preguntas relacionadas con la exposición materna a andrógenos o medicamentos y signos de virilización en la madre durante el embarazo.
Exploración física
Aunque es posible que diferentes trastornos se presenten con hallazgos parecidos durante la exploración física, a menudo existen aspectos del examen que son cruciales y que ayudan a guiar las pruebas iniciales.
Examen general: dado que algunas causas de los trastornos del desarrollo sexual (TDS) se asocian a insuficiencia suprarrenal, es importante confirmar que los signos vitales (presión arterial, llenado capilar y frecuencia cardíaca) y la glucemia son normales con organización de una monitorización adecuada. La presencia de rasgos dismórficos y/u otras anomalías congénitas pueden sugerir un síndrome que incluye genitales ambiguos (p. ej., el síndrome de Smith-Lemli-Opitz).
Gónadas: la presencia de una o dos gónadas descarta TDS 46, XX, secundarios a la deficiencia de 21 hidroxilasa. El TDS 46,XX ovotesticular se puede presentar raramente con gónadas inguinales. La presencia de dos gónadas se debe con mayor probabilidad a un TDS 46,XY. La presencia de una sola gónada palpable también podría sugerir un diagnóstico de disgenesia gonadal mixta.
Longitud de falo: para un bebé masculino a término, una longitud normal del pene estirado oscila de 2.5 cm a 4.5 cm y para un bebé femenino a término, una longitud del clítoris normal oscila de 0.2 cm a 0.85 cm con variación dentro de los grupos étnicos.[21]
Los neonatos con TDS 46,XX debidos a deficiencia de 21 hidroxilasa pueden presentar pliegues labioescrotales hiperpigmentados.
La puntuación de la masculinización externa (PME) puede ser un complemento útil a la exploración física para guiar al médico local.[22][23] La puntuación de los genitales externos (PGE) es una alternativa neutra desde el punto de vista del género, que tiene un alto nivel de correlación con el sistema de puntuación original de PME.[8][24][Figure caption and citation for the preceding image starts]: Puntuación de masculinización externaCreado por BMJ Knowledge Centre basado en Davies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. 2017 April [epub ahead of print]. [Citation ends].
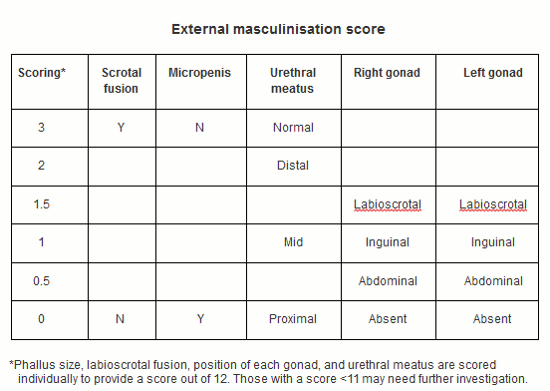
Orificio uretral: el hipospadias ocurre cuando no hay presente una abertura uretral en la punta del pene. Hipospadias asociado a separación de bolsas escrotales o testículos no descendidos puede sugerir un trastorno del desarrollo sexual subyacente. Si la abertura uretral se encuentra en la base del pene, se podría tratar de un seno urogenital en una mujer virilizada. Esto ocurre cuando las aberturas uretrales y vaginales están conectadas internamente y salen en el perineo a través de una abertura común.
Otros signos a observar son:[21]
Defectos de línea media (p. ej., paladar hendido)
Exceso de pigmentación (p. ej., pigmentación areolar)
Anormalidades esqueléticas o cardíacas.
Debe considerarse la posibilidad de realizar una prueba diagnóstica adicional del TDS en los bebés con micropene aislado, clitoromegalia aislada, hipospadias perineal aislado o cualquier hipospadias familiar, testículos no descendidos bilaterales aislados y los que presentan una EMA inferior a 11 (o una PGE inferior a 10.5). La presencia de un trastorno metabólico, de rasgos dismórficos o de antecedentes familiares relevantes debería reducir el umbral para una prueba diagnóstica más profunda.[8]
Presentaciones clínicas clave
Gónadas bilaterales impalpables en un bebé masculino aparente
Un neonato con gónadas bilaterales impalpables con un fenotipo masculino externo puede tener de hecho un cariotipo 46,XX con agrandamiento del clítoris debido a la producción excesiva de andrógenos en el útero. A estos bebés no se les debe dar de alta hasta que se haya excluido la hiperplasia suprarrenal congénita secundaria a deficiencia de 21 hidroxilasa.
Hernias inguinales en un bebé femenino aparente
Aunque un neonato pueda tener un cariotipo 46,XY subyacente, esta presentación puede ocurrir, por ejemplo en la insensibilidad completa del andrógeno, deficiencia de 17 beta-hidroxiesteroide deshidrogenasa y deficiencia de la 5 alfa-reductasa.
Genitales asimétricos
El mosaicismo del cromosoma sexual (p. ej., 45,X/46,XY) puede causar genitales asimétricos. En la laparoscopia, una gónada fibrosa y un hemiútero se identifican típicamente en el lado contralateral a la gónada descendida o palpable.
Micropene
Para un neonato varón nacido a término, la longitud normal del pene estirado oscila entre 2.5 cm y 4.5 cm.[21] La longitud <2.5 cm se considera anormal en un bebé varón nacido a término, particularmente ante la presencia de testículos no descendidos u otras alteraciones.[21] Hay variación en el tamaño normal del pene entre los grupos étnicos.
En neonatos varones prematuros, el pene es más corto y la longitud debe ser trazada en una tabla de percentil disponible para bebés prematuros.[25]
Los trastornos de la biosíntesis de la testosterona o la acción de la testosterona pueden llevar a un micropene (p. ej., insensibilidad androgénica parcia y deficiencia de 5 alfa-reductasa).
Agrandamiento del clítoris
La longitud normal del clítoris en una neonata a término oscila entre 0.2 cm y 0.85 cm, con variaciones entre distintos grupos étnicos.[21] La hiperplasia suprarrenal congénita secundaria a deficiencia de 21 hidroxilasa es la causa más común de cliteromegalia.
Hipospadias
La hipospadia grave puede estar asociada a genitales atípicos. La hipospadia grave puede presentar una causa genética subyacente en el 40% de los casos, incluidos la insensibilidad parcial a los andrógenos y la deficiencia de 5 alfa reductasa.[22][26] El neonato con hipospadias pero con gónadas impalpables tiene hiperplasia suprarrenal congénita secundaria a deficiencia de la 21 hidroxilasa hasta que se demuestre lo contrario.
Abordaje de un paciente con genitales atípicos
Después de realizar una anamnesis y exploración detalladas (que debe incluir la palpación de gónadas), el siguiente paso es organizar las pruebas diagnósticas específicas. En la presentación inicial, deben realizarse pruebas de control regulares de electrolitos, concentraciones de glucosa antes de comida y la presión arterial regular. Debe solicitarse un análisis urgente de cromosomas (cariotipo), con estudios por imágenes en las primeras 48 horas y estudios por hormonas tras las 48 horas de edad. En aquellos sin gónadas palpables, deben realizarse una 17-OH y otros estudios hormonales tras las 48 horas de edad. Un ultrasonido es útil para definir la presencia de un útero (estructura mülleriana), presencia de glándulas suprarrenales y la morfología de las gónadas. Debe solicitarse también una razón proteína-creatinina en orina.
Análisis cromosómico
En la evaluación inicial se debe realizar un análisis cromosómico con al menos 30 metafases para evaluar para el mosaicismo. Generalmente, los resultados se pueden obtener en 72 horas. Realizar un cariotipo formal puede tomar muchos días y se puede obtener un resultado rápido solicitando una hibridación fluorescente in situ (FISH) urgente o una reacción en cadena de la polimerasa (PCR) para la región de determinación del sexo Y (SRY). Si se considera un diagnóstico específico, se pueden realizar otros estudios genéticos.[9]
Los ensayos de secuenciación de alto rendimiento (SAR) y la secuenciación del genoma completo o del exoma (SGC) son métodos que se utilizan cada vez más para localizar múltiples genes relacionados con la TDS en un único análisis. Debe proporcionarse asesoramiento genético a los padres antes de las pruebas.[8]
Estudios por imágenes
Se realiza un ultrasonido pélvico y abdominal para determinar si existe un útero (una estructura Mülleriana). En un bebé, los estrógenos maternos aumentan la posibilidad de observar el útero a través de un ultrasonido durante las primeras semanas de vida. A menudo, los ovarios y las trompas de Falopio no son visibles en el ultrasonido. Un ultrasonido pélvico y abdominal también es útil para detectar la anatomía renal, anatomía suprarrenal (dado que las glándulas suprarrenales son relativamente grandes en el período neonatal), sitio de las gónadas y morfología.[27]
Estudios hormonales
Estas pruebas diagnósticas se realizan generalmente después de 48 horas de edad. Aunque ciertas presentaciones clínicas de los órganos genitales atípicos hacen algunos diagnósticos más probables, el médico debe tener en cuenta que un diagnóstico definitivo no se hace solamente con una exploración física, ya que hay presentaciones variadas en muchos trastornos del desarrollo sexual.
Sin gónadas palpables
En ausencia de gónadas palpables, debe excluirse una hiperplasia suprarrenal congénita secundaria a deficiencia de 21 hidroxilasa, ya que es el diagnóstico más probable.
Se deben obtener los niveles de 17 hidroxiprogesterona (17-OHP), los cuales serán considerablemente elevados en la deficiencia de 21 hidroxilasa.
En la deficiencia de 21 hidroxilasa, se debe comprobar y vigilar estrechamente los electrolitos, ya que es posible la pérdida de sal pueda tardar unos días en desarrollarse, típicamente en la segunda semana de vida. La medición de renina y aldosterona ayuda a evaluar la actividad mineralocorticoide.
Si se sospecha una hiperplasia suprarrenal congénita (HSC) y los niveles de 17-OHP no están marcadamente aumentados, se debe continuar la evaluación de los precursores suprarrenales para buscar otras causas menos frecuentes de HSC, incluidas el 11-desoxicortisol y 11-desoxicorticosterona, con el fin de descartar la deficiencia de 11-beta hidroxilasa. Un perfil de esteroides en orina puede ayudar en este contexto.
Si el lactante requiere un tratamiento hormonal con esteroides de forma inmediata, deben recogerse muestras adecuadas de orina y suero antes de iniciar el tratamiento y guardarlas para su posterior análisis.[8]
Gónadas palpables
Testosterona y dihidrotestosterona (DHT): un alto ratio de testosterona frente a DHT sugiere una deficiencia de 5-alfa reductasa. Un ratio bajo de testosterona frente a androstenediona sugiere una deficiencia de 17-beta-hidroxiesteroide deshidrogenasa.
Hormona luteinizante (LH) y hormona foliculoestimulante (FSH): ambas ayudan a evaluar el eje hipotalámico-hipofisario-gonadal. Niveles bajos en la primera semana de vida no indican necesariamente hipogonadismo hipogonadotrópico; sin embargo, una evaluación entre los 2 y los 4 meses de vida puede ser más definitoria cuando existe un aumento fisiológico en los niveles de LH y FSH.
Prueba de estimulación con hormona adrenocorticotrópica (ACTH): esta prueba se utiliza en casos específicos para investigar anomalías de la síntesis de glucocorticoides en algunas formas de TDS.
Prueba de estimulación con gonadotrofina coriónica humana (hCG): evalúa la capacidad de las células de Leydig de los testículos para responder a la hCG (un análogo del receptor de la LH) y producir testosterona. El ratio entre testosterona frente a DHT después de la estimulación con hCG se utiliza para detectar la deficiencia de 5-alfa reductasa. La prueba de estimulación de la hCG también puede identificar un bloqueo en la vía biosintética de la testosterona, como en la deficiencia de 17-beta-hidroxiesteroide-deshidrogenasa, donde el ratio entre testosterona y androstenediona sería bajo.
Hormona antimülleriana (HAM) (antes denominada sustancia inhibidora de la mülleriana): puede obtenerse para evaluar la función testicular (células de Sertoli) en un bebé con sospecha de DSD 46,XY y en el TDS cromosómico. Los niveles de HAM dependen de la edad y del sexo.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad