Etiología
En el desarrollo normal, el desarrollo sexual está regido por dos procesos consecutivos: determinación del sexo seguida de la diferenciación del sexo. La determinación del sexo es la formación de un testículo o un ovario de una gónada bipotencial y es conducida por la expresión secuencial de un número de genes. La diferenciación del sexo es el desarrollo de características físicas internas y externas, provocada por la acción de la hormona gonadal en los tejidos diana.
[Figure caption and citation for the preceding image starts]: Ejemplos de genes que controlan diferentes etapas de la determinación del sexo; los genes determinantes ováricos son objeto de investigación en cursoDavies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. 2017;102(10):968-74. [Citation ends]. [Figure caption and citation for the preceding image starts]: Representación esquemática de la diferenciación de sexo. Glosario: 5 alfa-reductasa (5α-R), hormona antimülleriana (HAM), dihidrotestosterona (DHT)Davies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. 2017;102(10):968-74. [Citation ends].
[Figure caption and citation for the preceding image starts]: Representación esquemática de la diferenciación de sexo. Glosario: 5 alfa-reductasa (5α-R), hormona antimülleriana (HAM), dihidrotestosterona (DHT)Davies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. 2017;102(10):968-74. [Citation ends].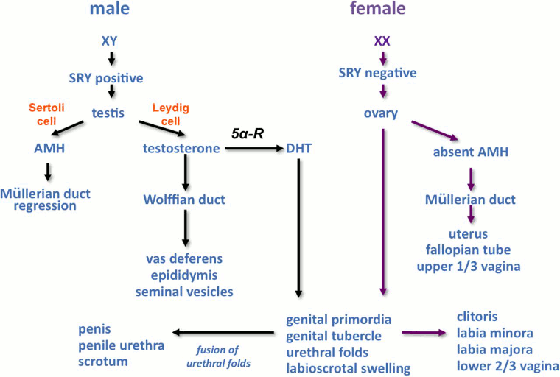 Ejemplos de genes que controlan las diferentes etapas de la determinación del sexo; los genes determinantes ováricos son objeto de investigación en curso.
Ejemplos de genes que controlan las diferentes etapas de la determinación del sexo; los genes determinantes ováricos son objeto de investigación en curso.
Representación esquemática de la diferenciación de sexo. Glosario: 5 alfa reductasa (5α-R), hormona antimülleriana (HAM), dihidrotestosterona (DHT).
Diversos trastornos con una base genética subyacente causan diferencias en el desarrollo del sexo (TDS) y se presentan con genitales atípicos en el neonato. Estos resultan de la determinación o diferenciación atípica del sexo. En algunas ocasiones no muy frecuentes, puede producirse la virilización de un feto 46,XX debido a tumores virilizantes maternos o a la exposición materna a fármacos androgénicos.
TDS de los cromosomas sexuales:
Los individuos pueden presentar genitales atípicos debido a la ganancia o pérdida meiótica o mitótica de un cromosoma sexual, o debido a mosaicismo (que es poco frecuente). La disgenesia gonadal mixta 45,X/46,XY se debe a errores posmeióticos, los cuales conducen a la pérdida de un cromosoma Y en un linaje celular. El mosaicismo 46,XX/46,XY es el resultado de la fusión de dos cigotos.
TDS 46,XY:
La disgenesia gonadal puede producirse debido a mutaciones en uno de varios genes que intervienen en el desarrollo testicular.[11] Los defectos en algunos de estos genes (p. ej., región de determinación del sexo Y [SRY]) también pueden provocar un trastorno del desarrollo sexual ovotesticular.
Los trastornos en la biosíntesis de andrógenos incluyen mutaciones en genes de la vía de la síntesis de testosterona que o bien son comunes a la glándula suprarrenal y a los testículos y, por lo tanto también pueden causar deficiencias de glucocorticoides y/o mineralocorticoides (con algunas formas de hiperplasia suprarrenal congénita [HSC]), o bien se encuentran únicamente en los testículos y por ello solo conducen a defectos en la biosíntesis de testosterona. Los ejemplos son la deficiencia de 17-alfa-hidroxilasa, 20-22 liasa debido a mutaciones en CYP17; deficiencia de 3-hidroxiesteroide deshidrogenasa (HSD) debida a mutaciones en HSD3B2, y la deficiencia de la proteína reguladora aguda esteroidogénica (StAR). Todas estas son enfermedades autosómicas recesivas.
La causa del síndrome de insensibilidad androgénica parcial son las mutaciones en el receptor androgénico en Xq11 y se trata de una enfermedad recesiva ligada al cromosoma X.[12]
El hipogonadismo hipogonadotrópico congénito se puede deber a uno de varios defectos de genes y puede ser aislado o estar asociado a otras deficiencias de hormonas hipofisarias.[13][14]
TDS 46,XX:
La hiperplasia suprarrenal congénita (HSC) clásica debida a la deficiencia de 21 hidroxilasa (CYP21A2 en el cromosoma 6p21.3) es la causa de los genitales atípicos en más del 95% de los casos. Esta es una enfermedad autosómica recesiva. Otras causas de TDS 46,XX son la deficiencia de 11-beta hidroxilasa (CYP11B1 en el cromosoma 8q21) y la deficiencia de 3-beta-HSD (HSD3B2).
Los trastornos del desarrollo sexual ovotesticulares pueden ser causados por una serie de defectos genéticos, incluida la translocación de una región de determinación del sexo Y (SRY) a un cromosoma X por duplicación de la región SOX9.[1]
Fisiopatología
Un ejemplo de trastorno del desarrollo sexual (TDS) de los cromosomas sexuales es la disgenesia gonadal mixta, que se caracteriza por el desarrollo asimétrico de las gónadas y conductos genitales internos. Existe un testículo disgenético y, por lo tanto, los conductos de Wolff están poco desarrollados en un lado y hay una gónada estriada (ovario) en el otro lado. Se observan grados variables de ambigüedad de los genitales externos. La falta de un cromosoma Y, y por ello, la falta del gen de la región determinante del sexo Y (SRY), por un lado conduce a disgenesia testicular que produce testosterona insuficiente para la virilización masculina normal. El grado de virilización depende del grado de funcionamiento de los testículos.
En el TDS 46,XY que se presenta con genitales atípicos, el grado de virilización depende de la cantidad de funcionamiento residual de los testículos, de la cantidad de testosterona sintetizada o del grado de insensibilidad androgéna.
En el TDS 46,XX con hiperplasia suprarrenal congénita debida a la deficiencia de 21 hidroxilasa, el grado de virilización depende de la actividad residual del gen 21 hidroxilasa. En ausencia de cualquier actividad de 21 hidroxilasa (mutaciones nulas en ambos alelos), los precursores de cortisol son trasladados a la producción de andrógenos, lo cual lleva a niveles de andrógenos altos y a virilización. Esto se asocia usualmente a la pérdida de sal.[15][Figure caption and citation for the preceding image starts]: Ejemplos seleccionados de producción anormal de testosterona (rojo) o de acción de la testosterona (púrpura) en las diferencias de desarrollo sexual 46,XY. Glosario: deficiencia de 3 beta-hidroxiesteroide deshidrogenasa (3β-HSD), deficiencia de 17 alfa-hidroxilasa (17α-HSD), deficiencia de 17 beta-hidroxiesteroide deshidrogenasa (17β-HSD), receptor androgéno (RA), insensibilidad androgéna completa, insensibilidad androgéna parcial (IAP)Davies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. 2017 April [epub ahead of print]. [Citation ends].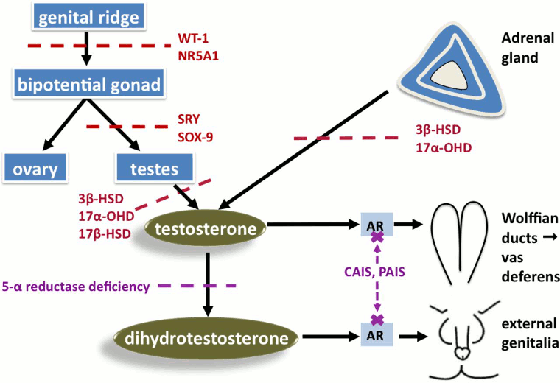 [Figure caption and citation for the preceding image starts]: Ejemplos seleccionados de causas de exceso de andrógenos en las diferencias de desarrollo sexual 46,XXDavies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. 2017 April [epub ahead of print]. [Citation ends].
[Figure caption and citation for the preceding image starts]: Ejemplos seleccionados de causas de exceso de andrógenos en las diferencias de desarrollo sexual 46,XXDavies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. 2017 April [epub ahead of print]. [Citation ends].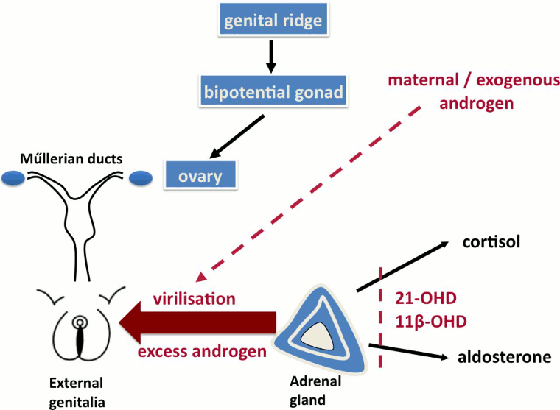
Clasificación
Declaración de consenso sobre el manejo de los trastornos de intersexualidad[3]
Trastornos del desarrollo sexual (TDS) de los cromosomas sexuales: no es el cariotipo típico 46,XX ni 46,XY.
TDS 46,XY: cromosómicamente masculino, pero el TDS resulta de:
Un defecto en el desarrollo testicular.
Un trastorno en la síntesis o la acción de los andrógenos.
Síndromes asociados a defectos en el desarrollo de los genitales, que incluyen alteraciones cloacales, otros síndromes genéticos, síndrome de los testículos en ascenso e hipogonadismo hipogonadotrópico congénito.
TDS 46,XX: cromosómicamente femenino, pero el TDS resulta de:
Defectos en el desarrollo ovárico.
Trastornos que conducen a niveles altos de andrógenos; la hiperplasia suprarrenal congénita es, de lejos, la causa más frecuente.
Alteraciones de los conductos de Müller (disgenesia o hipoplasia), útero o vagina.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad