Etiología
La mayoría de los CCR se producen de forma esporádica. El tabaquismo es el factor de riesgo mejor establecido.[27][30] El aumento de la edad, la obesidad y la hipertensión se asocian a un mayor riesgo.[19][27][30] Se han implicado el trasplante renal, la enfermedad renal terminal con diálisis y la exposición a radiación pélvica.[26][31][32]
Se han identificado varios síndromes genéticos familiares.[30] Entre los genes relacionados con el cáncer de riñón se encuentran Von Hippel-Lindau (VHL), foliculina (FLCN), succinato deshidrogenasa (SDH), factor de transición mesenquimal-epitelial (MET), fumarato hidratasa (FH) y BAP1 (codifica para la proteína-1 asociada a BRCA1 [ubiquitina carboxi-terminal hidrolasa]), que se heredan de forma autosómica dominante.[33] La identificación de las causas genéticas del CCR tiene consecuencias para la vigilancia y el uso de terapias dirigidas.
El síndrome de VHL se asocia con la variante de células claras del CCR. El riesgo de por vida de CCR en el síndrome de VHL es del 70%.[33] La evidencia sugiere que entre el 60% y el 70% de los pacientes con CCR de células claras esporádico también tiene mutaciones en VHL.[34] Los pacientes con el síndrome de VHL presentan una inactivación de la línea germinal de un alelo del VHL (que se encuentra en el cromosoma 3p); una segunda mutación de pérdida de funcionalidad es un evento desencadenante precoz para cánceres VHL, como el CCR. Los productos del gen VHL actúan como supresores de tumor y son factores fundamentales en el desarrollo del CCR de células claras. El CCR se desarrollará entre la segunda y la cuarta décadas de la vida en el 25% al 45% de los pacientes con síndrome VHL. Los cánceres son más a menudo bilaterales y/o multifocales; sin embargo, el CCR en el síndrome VHL suele tener un curso indolente, y los tumores <3 cm en estos pacientes raramente metastatizan.[35] La vigilancia periódica del CCR en pacientes con síndrome de VHL está justificada.[33]
El síndrome de Birt-Hogg-Dubé está causado por una mutación en la línea germinal del gen FLCN. Se asocia predominantemente a los cánceres renales cromófobos y oncocíticos, pero incluye otros subtipos, como el CCR de células claras. Los pacientes con una mutación de la FLCN tienen un riesgo estimado de CCR de aproximadamente un 25%.[33]
El CCR relacionado con la succinato deshidrogenasa presenta varios tipos histológicos, que incluyen células claras, y está asociado con mutaciones de estirpe germinal en cada una de las subunidades SDH, siendo el SDHB el gen más comúnmente asociado.[33] El riesgo de por vida de CCR en portadores de la mutación SDHB es probablemente menos del 10% al 15%.[33] Las mutaciones de estirpe germinal en SDHB pueden presentarse con un fenotipo familiar de CCR solo, pero las mutaciones de los genes que codifican las subunidades de la succinato deshidrogenasa están asociadas con feocromocitomas y paragangliomas, y con tumores del estroma gastrointestinal.[33][36] La vigilancia renal mediante estudios por imágenes de resonancia magnética puede combinarse con la detección del feocromocitoma/paraganglioma en este grupo de pacientes.[33]
Otras histologías del CCR no muestran predominancia de mutaciones en VHL. Por ejemplo, el CCR papilar de tipo 1 presenta principalmente mutaciones en la proteína c-MET, mientras que el tipo 2 (como parte de la leiomiomatosis hereditaria y el cáncer de células renales) presenta mutaciones en el gen FH.[33]
La esclerosis tuberosa puede estar asociada con el CCR de inicio temprano, pero el CCR es poco frecuente en esta afección y las lesiones renales son más frecuentemente angiomiolipomas.[33]
Fisiopatología
Los diversos subtipos de carcinoma de células renales (CCR) están asociados a distintas alteraciones citogenéticas. Los cánceres con cambios genéticos diferentes tienen histología y significación clínica diferentes.[33][36]
La mayoría de los CCR tienen una histología de células claras, una minoría tiene una histología papilar o cromófoba:[37]
Carcinomas de células claras
Representan entre el 70% y el 90% de los CCR.
Suelen ser lesiones corticales solitarias, pero en el síndrome de von Hippel-Lindau se observan casos bilaterales (también pueden ser múltiples).
Suele ser de color amarillo dorado en la patología macroscópica; puede mostrar degeneración quística, hemorragia, cambios sarcomatoides y extensión a la vena renal; el grado del nucleolo es el factor pronóstico más importante después de la etapa clínica.
Pronóstico globalmente peor que el subtipo papilar o cromófobo, pero mayor respuesta al tratamiento sistémico; los casos esporádicos muestran a menudo pérdida del cromosoma 3p.
Se observan mutaciones de von Hippel-Lindau (VHL), tanto en casos esporádicos, como en aquellos asociados con el conocido síndrome de VHL.
CCR papilar
Representan entre el 10% y el 15% de los CCR.
Más comúnmente bilateral, o multifocal con grado variable de papilas.
Presentan hemorragias y necrosis frecuentes.
En general, pueden tener mejor pronóstico que los carcinomas de células claras.
Asociada a trisomía 7, 17 y pérdida de Y.
Tumores cromófobos
Representan entre el 3% y el 5% de los CCR.
Células grandes y pálidas.
Por lo general, curso relativamente indolente de la enfermedad.
El conocimiento de la fisiopatología molecular del CCR de células claras ha aumentado gracias al análisis de síndromes hereditarios de CCR como el VHL.[33][35]La proteína VHL funciona principalmente como ubiquitina ligasa; uno de sus principales objetivos de su degradación es el factor inducible por hipoxia (FIH). El FIH promueve la transcripción de múltiples genes relacionados con la hipoxia, que se activan constitutivamente en ausencia del VHL. Estos genes incluyen el factor de crecimiento endotelial vascular (VEGF), que promueve la angiogénesis; el factor de crecimiento derivado de plaquetas (PDGF), conocido oncogén; los receptores del factor de crecimiento epidérmico, que promueve el crecimiento celular; y las metaloproteinasas de matriz, que son parte integral de la invasión tumoral. Estos genes controlados por el FIH (especialmente el VEGF) han sido objeto de varias estrategias de tratamiento molecular. Los fármacos que se utilizan comúnmente en el tratamiento del CCR son los inhibidores de la tirosina cinasa, diseñados para inhibir las vías de los receptores del VEGF y del PDGF. Las proteínas cinasas críticas para la angiogénesis están sobreexpresadas o sobreactivas en los tumores. Inhibir la función cinasa en las células tumorales inhibe su interacción con el microambiente, incluida la neovasculatura, lo que tiene efectos anticáncer y tóxicos.[38] La misma transcripción del FIH es controlada por la vía de la PI3 cinasa, de la cual, el blanco de la rapamicina en mamíferos (m-TOR) tiene un papel clave en la regulación ascendente y, por ello, la m-TOR también es objetivo terapéutico en el CCR actualmente. Otros objetivos de las células VHL incluyen el p53, determinadas polimerasas del ARN y la fibronectina. Luego de la pérdida inicial de los alelos del VHL, como parte del síndrome VHL o de forma esporádica, es probable que las acumulaciones de otros defectos genéticos no aleatorios contribuyan al desarrollo tumoral.
El microambiente inmunológico es otro factor objetivo en el área terapéutica del cáncer. Para evitar la autoinmunidad, el organismo cuenta con proteínas de punto de control inherentes, como la proteína asociada a linfocitos T citotóxicos 4 (CTLA-4) y el ligando 1 de muerte celular programada (PD-L1); sin embargo, estas pueden ser usadas por células cancerosas para evitar la detección por parte del sistema inmunitario.[38] Muchas células tumorales expresan CTLA-4 y/o PD-L1, que se vinculan a la proteína de muerte celular programada 1, e inhiben eficazmente a las células T de atacar a las células tumorales.[38] Los anticuerpos monoclonales dirigidos contra estas proteínas relacionadas con la muerte celular impiden la activación de dichas señales inmunosupresoras y superan esta tolerancia inmunológica, lo que permite un efecto anticanceroso de origen inmunológico.[38]
La exposición al tabaco favorece el desarrollo del CCR.[39][40] Los mecanismos biológicos pueden incluir el estrés oxidativo y la exposición a las nitrosaminas y otros carcinógenos en el humo del tabaco. Se ha demostrado que la nicotina estimula la angiogénesis patológica y acelera el crecimiento tumoral.[30]
Algunos de los mecanismos propuestos del CCR inducido por obesidad son el incremento de la peroxidación lipídica que forma aductos de ADN, el incremento del factor de crecimiento similar a la insulina y mayores tasas de filtración glomerular y nefroesclerosis que causan un incremento en la exposición a carcinógenos.[26][30][41]
El mecanismo propuesto del CCR inducido por hipertensión es el daño del túbulo y un aumento en la exposición a carcinógenos.[26][30]
Clasificación
Tamaño de la masa renal
Se define a la masa renal pequeña (MRP) como una lesión renal <4 cm (o 3 cm según algunas autoridades) que muestra posibles características de un carcinoma de células renales (CCR) (con un aumento de tamaño anormal) en los estudios por imágenes. Es más probable que las MRP (particularmente aquellas <2 cm) sean benignas (hasta el 46% de aquellas <1 cm y el 25% de aquellas <2 cm).[3][4][5] Las masas de <3.5 cm (aun siendo CCR) tienen un bajo potencial metastásico a lo largo de 2 y 3 años. Las masas que alcanzan los 4 cm y/o aumentan de tamaño 5 mm en 12 meses pueden requerir intervención.[6]
Sistema de estadificación según tumor, ganglio y metástasis (TNM) del CCR[7][8]
La clasificación de tumor, ganglio y metástasis (TMN) de los tumores malignos describe la extensión de la enfermedad basándose en los siguientes factores anatómicos:
Tamaño y extensión del tumor primario (T)
Afectación de los ganglios linfáticos regionales (N); y
Presencia o ausencia de metástasis a distancia (M).
Estadificación
El CCR de etapa temprana (etapas 1 y 2) se define como un tumor confinado al riñón sin nódulos linfáticos regionales o metástasis distantes.
Los tumores en etapa 3 se extienden dentro de las venas principales o invaden la glándula suprarrenal o el tejido perinéfrico sin atravesar la fascia de Gerota. Puede haber metástasis en un solo ganglio linfático regional pero sin evidencia de metástasis a distancia.
Los tumores de la etapa 4 se extienden más allá de la fascia de Gerota o tienen metástasis distantes.
Clasificación de la Organización Mundial de la Salud (OMS): tumores de células renales[9]
La clasificación de la OMS de 2022 introdujo una clasificación de tumores renales basada en datos moleculares, además de los tumores renales basados en la morfología.[10]
Tumores renales de células claras
Carcinoma de células renales de células claras (CCR)
Neoplasia renal quística multilocular de bajo potencial maligno
Tumores renales papilares
Adenoma papilar
CCR papilar (ya no se divide en tipo I y II)
Tumores renales oncocíticos y cromófobos
Oncocitoma de riñón
CCR cromófobo
Otros tumores oncocíticos del riñón
Tumores del conducto colector
Carcinoma del túbulo colector
Otros tumores renales
Tumor renal papilar de células claras
Carcinoma mucinoso tubular y de células fusiformes
CCR tubulocístico
CCR quístico asociado a enfermedad adquirida
CCR sólido y quístico eosinofílico (ESC)
CCR NOS (no especificado)
Tumores renales definidos molecularmente
CCR reordenados por TFE3
CCR alterado por TFEB (CCR reordenado por TFEB y CCR amplificado por TFEB)
CCR con mutación ELOC (antes TCEB1)
CCR con deficiencia de fumarato hidratasa
CCR con deficiencia de succinato deshidrogenasa
CCR con ALK
Carcinoma medular renal deficiente en SMARCB1
Organización Mundial de la Salud (OMS)/International Society of Urological Pathology (ISUP): clasificación[11][12]
La prominencia nucleolar define los grados 1-3 de los CCR de células claras y papilares. El pleomorfismo nuclear extremo, o diferenciación sarcomatoide y/o rabdoide, define los tumores de grado 4.
Grado 1: núcleos de células tumorales ausentes o inconspicuos y basófilos con un aumento de 400 veces.
Grado 2: núcleos de células tumorales conspicuos y eosinófilos con un aumento de 400 veces y visibles, pero no prominentes con un aumento de 100 veces.
Grado 3: núcleos de células tumorales conspicuos y eosinófilos con un aumento de 100 veces
Grado 4: tumores que muestran pleomorfismo nuclear extremo, células gigantes tumorales y/o la presencia de alguna proporción del tumor que muestre una desdiferenciación sarcomatoide o rabdoide.
Clasificación de Bosniak
La clasificación de Bosniak fue diseñada para clasificar las masas quísticas renales en cinco categorías según las técnicas radiológicas de TC e IRM.[13][14][15]
[Figure caption and citation for the preceding image starts]: Clasificación de BosniakTabla creada por el Dr. Rodrigo R. Pessoa MD; usada con permiso [Citation ends].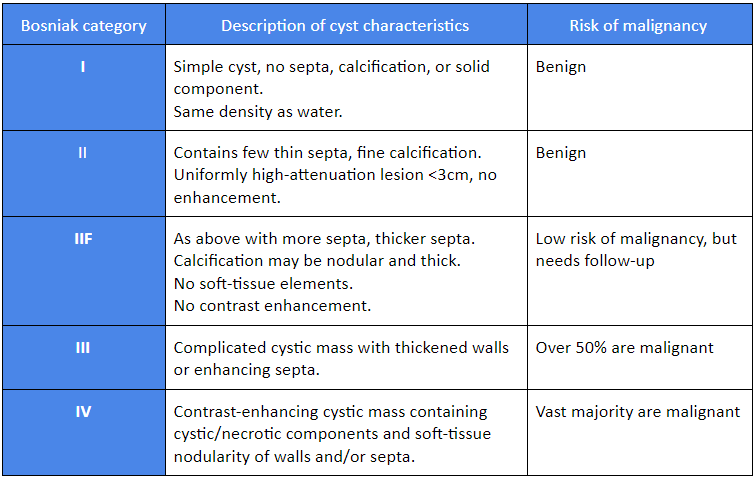
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad