Anamnesis y examen
Principales factores de diagnóstico
común
presencia de factores de riesgo
Entre los factores de riesgo importantes se incluyen los antecedentes familiares de retraso puberal (puede ser específico del retraso constitucional o del hipogonadismo hipogonadotrófico orgánico), las anomalías hipofisarias congénitas, las mutaciones genéticas conocidas, los trastornos cromosómicos, un diagnóstico sindrómico, la anosmia, los trastornos alimentarios, las enfermedades sistémicas crónicas, la desnutrición, el ejercicio intenso y las anomalías gonadales congénitas y adquiridas.
niños: testículos <4 ml
El tamaño testicular se documenta a través de la medición del eje más largo o por el volumen testicular utilizando el orquidómetro de Prader.
[Figure caption and citation for the preceding image starts]: Orquidómetro de PraderCreado por BMJ Knowledge Centre [Citation ends]. [Figure caption and citation for the preceding image starts]: Método para comparar el tamaño testicular utilizando el orquidómetro de PraderDe la colección del Dr. A. Mehta [Citation ends].
[Figure caption and citation for the preceding image starts]: Método para comparar el tamaño testicular utilizando el orquidómetro de PraderDe la colección del Dr. A. Mehta [Citation ends].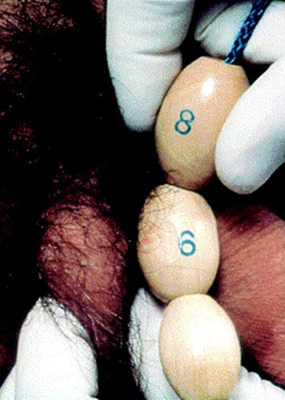 Un volumen de 4 mL o una longitud de 2.5 cm define el inicio de la pubertad.[39]f
Un volumen de 4 mL o una longitud de 2.5 cm define el inicio de la pubertad.[39]f
niñas: ausencia de desarrollo mamario
El primer signo demostrable de pubertad en niñas es el desarrollo mamario.[38]
ausencia de vello púbico/axilar
El vello púbico y axilar, el acné y el olor corporal se desarrollan como resultado de andrógenos secretados por la glándula suprarrenal. El inicio del vello axilar ocurre en la etapa media de la pubertad.
ausencia de menarquia >3 años desde el brote de la mama
La menarca se utiliza como evidencia documentada y bien definida de pubertad en niñas.
Se presenta típicamente junto con el desarrollo mamario del estadio 4 de Tanner en la mayoría de las niñas.
ausencia de brote de crecimiento
Las hormonas sexuales estimulan directamente la placa de crecimiento, lo que produce el brote de crecimiento. También hay un aumento en la secreción de la hormona del crecimiento (GH).[33] El estrógeno ya sea del ovario o aromatizado a partir de la testosterona testicular es el factor que media la mayor respuesta a la GH durante la pubertad.[34]
El brote de crecimiento ocurre en la etapa media-final de la pubertad en niños y en la etapa 3 del desarrollo mamario en niñas.
Por término medio, aporta 25 cm de altura en las hembras y 30 cm en los machos.
anosmia
El síndrome de Kallman se asocia con hipogonadismo hipogonadotrópico orgánico y nervios olfativos hipoplásicos, lo que resulta en anosmia.[21] Otros signos clave asociados al hipogonadismo hipogonadotrófico orgánico (y conocidos como "señales de alarma" de esta afección) son la criptorquidia bilateral, micropene (2 o más desviaciones estándar por debajo de la longitud media para la edad), defectos de la línea media como labio leporino, paladar hendido y agenesia renal, así como la sincinesia (movimientos en espejo) que es patognomónica de esta afección.
Otros factores de diagnóstico
común
estatura baja
El retraso constitucional y el síndrome de Turner están asociados con la estatura baja.
infrecuente
rasgos dismórficos
Las niñas que tienen el síndrome de Turner son bajas con varios rasgos dismórficos que incluyen línea de implante capilar posterior baja, cuello alado y orejas prominentes rotadas hacia atrás. Las características al momento de la presentación pueden ser más sutiles y deben realizarse investigaciones para descartar el síndrome de Turner en las niñas que tienen un retraso de la pubertad.[40]
Se debe evaluar a los pacientes en busca de desproporción corporal. Los niños que tienen el síndrome de Klinefelter tienen estatura alta con mayor longitud de las extremidades inferiores. Además, tienen testículos disgenéticos y un retraso en el desarrollo y pueden tener ginecomastia.[27]
Pueden existir otros rasgos que sugieran un diagnóstico sindrómico (p. ej., Prader-Willi, Bardet-Biedl, CHARGE, o displasia septoóptica).
Factores de riesgo
Fuerte
antecedentes familiares de retraso puberal
El retraso constitucional de la pubertad, la causa más común de retraso puberal, tanto en hombres como en mujeres, se agrupa en familias y suele mostrar un patrón de herencia autosómico dominante.[1][19][20] Entre el 50% y el 75% de las personas con retraso constitucional presenta antecedentes familiares de retraso puberal.[1]
Existe una similitud en la edad a la pubertad entre las niñas y sus madres, especialmente con la edad de la menarquia.
anomalías estructurales hipofisarias congénitas
Puede ser por mutaciones genéticas o anomalías estructurales dentro del eje hipotalámico-hipofisario asociadas con defectos de la línea media del prosencéfalo (p. ej., displasia septoóptica u holoprosencefalia).[22] La deficiencia resultante de gonadotropina puede ser aislada o combinada con otras deficiencias de hormonas hipofisarias.
mutaciones genéticas
Varios genes se han relacionado con la patogénesis del hipogonadismo hipogonadotrófico, como los genes ANOS1, FGFR1, KISS1R, KISS-1, GNRHR, PROK2, PROKR2, NSMF, NROB1, subunidad beta de LH y FSH, leptina, receptor de leptina y prohormona convertasa 1 (PC1).[24]
enfermedades cromosómicas
El síndrome de Klinefelter (XXY), el síndrome de Turner (45X), la disgenesia gonadal XY y la disgenesia gonadal mixta 45X/46XY están asociados con gónadas disgenéticas e hipogonadismo hipergonadotrópico.[27] Los hombres con síndrome de Klinefelter suelen entrar en la pubertad pero no llegan a progresar del todo, con concentraciones crecientes de hormona foliculoestimulante y concentraciones decrecientes de testosterona desde mediados de la pubertad.
diagnóstico sindrómico
Los síndromes de Prader-Willi, Bardet-Biedl y CHARGE están asociados con el hipogonadismo hipogonadotrópico.
alimentación restrictiva
La anorexia nerviosa y los patrones alimentarios anormales pueden provocar hipogonadismo hipogonadotrófico.
enfermedad sistémica crónica
Afecciones comunes como la cardiopatía crónica, el asma de moderada a grave, la fibrosis quística, la enfermedad celíaca, la enfermedad inflamatoria intestinal (enfermedad de Crohn y colitis ulcerosa), los trastornos inflamatorios (p. ej., la artritis idiopática juvenil), la insuficiencia renal crónica, cualquier neoplasia maligna crónica y la diabetes mellitus mal controlada pueden provocar hipogonadismo hipogonadotrófico funcional.
desnutrición
Puede provocar hipogonadismo hipogonadotrófico funcional.
ejercicio intenso
El ejercicio físico intenso puede provocar el retraso del inicio de la pubertad, el retraso de la menarquia o la detención del desarrollo puberal.
anomalías testiculares congénitas
El hipogonadismo hipergonadotrófico puede deberse a la anorquia o a la regresión testicular en los hombres. La criptorquidia bilateral grave (testículos no descendidos) también puede asociarse a anomalías testiculares congénitas.
anomalías de la diferenciación gonadal adquiridas
El hipogonadismo hipergonadotrófico puede deberse a una torsión testicular o a un tumor.
La endocrinopatía autoinmunitaria puede provocar daños gonadales e hipogonadismo hipergonadotrófico.[29]
cirugía de la hipófisis
Puede provocar (p. ej., tras la extracción de un craneofaringioma o un adenoma) hipogonadismo hipogonadotrópico.[25]
hipoplasia suprarrenal
Las mutaciones de NROB1 causan hipogonadismo hipogonadotrópico e hipoplasia suprarrenal congénita que puede provocar una crisis suprarrenal neonatal grave. Otras mutaciones implicadas en la hipoplasia suprarrenal congénita son las de STAR, CYP11A1 y CYP17A1.[35] Esta afección se hereda como un trastorno ligado al cromosoma X.[23]
Débil
quimioterapia
Puede provocar hipogonadismo hipogonadotrópico o hipergonadotrópico según el agente quimioterapéutico utilizado. El riesgo para la quimioterapia es mayor con los fármacos alquilantes.[36]
radioterapia
Puede provocar hipogonadismo hipogonadotrópico en la radiación hipofisaria/cerebral o hipogonadismo hipergonadotrópico posterior a la radiación de la pelvis.[30]
histiocitosis
Puede provocar hipogonadismo hipogonadotrópico permanente.
anemia falciforme
Puede provocar hipogonadismo hipogonadotrópico permanente.
sobrecarga de hierro (asociada con transfusiones)
Puede provocar hipogonadismo hipogonadotrópico permanente.[26]
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad