Etiología
La mayoría de los pacientes que presentan un retraso puberal tienen un retraso temporal debido a una inhibición central del eje hipotálamo-hipofisario que se resuelve con el tiempo y la observación seriada o un tratamiento breve con esteroides sexuales (retraso funcional).
El retraso constitucional (también llamado autolimitado) es la causa más frecuente de retraso puberal tanto en hombres como en mujeres.[1][19] Suele presentarse en familias (del 50% al 75% de los casos) y se asocia a una estatura baja adecuada a la edad ósea.[1][19][20] Puede ser difícil de distinguir de la deficiencia orgánica de gonadotrofinas.
También puede producirse un retraso puberal temporal o reversible debido a cualquier enfermedad crónica infantil (p. ej., fibrosis quística, enfermedad inflamatoria intestinal, enfermedad celíaca, diabetes mellitus mal controlada), ejercicio físico excesivo, malnutrición y trastornos alimentarios (anorexia nerviosa).
Las causas orgánicas pueden ser por una causa hipotalámico-hipofisaria o por una anomalía de la diferenciación gonadal.
Trastornos hipotalámico-hipofisarios
El hipogonadismo hipogonadotrófico puede ser congénito o adquirido, y puede ser aislado o estar asociado a deficiencias hormonales hipofisarias combinadas. Si se asocia al fenotipo de anosmia, se denomina síndrome de Kallmann.[21] También puede asociarse a trastornos cerebrales de la línea media en la displasia septoóptica y la holoprosencefalia, a hipoplasia suprarrenal o a otros síndromes como los de Prader-Willi, Bardet-Biedl y CHARGE.[22][23] Del mismo modo, se observan formas parciales de hipogonadismo hipogonadotrófico, a veces con una entrada en la pubertad normalmente programada seguida de una detención puberal, y éstas pueden ser particularmente difíciles de distinguir del retraso constitucional (autolimitada) de la pubertad.
Se han relacionado más de 50 genes con la patogénesis del hipogonadismo hipogonadotrófico congénito, incluidos los genes ANOS1, FGFR1, KISS1R, KISS-1, GNRHR, PROK2, PROKR2, NSMF, NROB1, la subunidad beta de la LH y la FSH, la leptina, el receptor de la leptina y la prohormona convertasa 1 (PC1).[24]
Las formas adquiridas de hipogonadismo hipogonadotrófico se asocian a traumatismos intracraneales, tumores, cirugía o radioterapia.[25] La histiocitosis, la anemia falciforme y la sobrecarga de hierro (asociada con transfusiones) pueden producir deficiencia de gonadotropina permanente.[26]
Trastornos gonadales
Los signos clínicos de trastornos congénitos incluyen criptorquidia/anorquia.
Los trastornos cromosómicos incluyen el síndrome de Klinefelter (XXY), el síndrome de Turner (45X), la disgenesia gonadal XY y la disgenesia gonadal mixta 45X/46XY.[27][28] La insuficiencia ovárica primaria se ha asociado a la deficiencia de varios genes subyacentes, como FMR1, FMR2, BMP15, DIAPH2 y FOXL2.
Las causas adquiridas incluyen torsión testicular, enfermedades autoinmunitarias poliglandulares, quimioterapia, infecciones virales (p. ej. paperas), radiación pélvica o abdominal, y cirugía testicular u ovárica.[29][30]
Fisiopatología
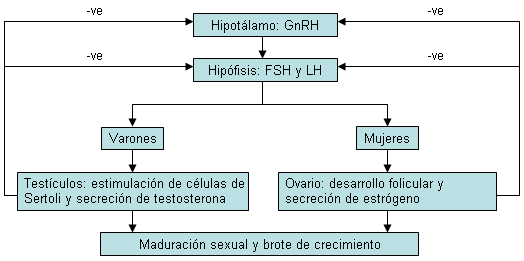
El inicio de la pubertad está controlado por varios factores.[6] El aumento nocturno de la amplitud y la pulsatilidad de la secreción de la hormona liberadora de gonadotrofina (GnRH) por el hipotálamo es la primera prueba de la activación puberal del eje hipotalámico-hipofisario-gonadal.[31] Otros neurotransmisores (p. ej. la acetilcolina, el ácido gamma-aminobutírico, los péptidos opioides, las prostaglandinas y la serotonina) y la leptina pueden modular el inicio de la pubertad. El KISS1R es un receptor acoplado a proteína G que es un ligando para la kisspeptina, y la estimulación del receptor kisspeptina-1 (localizado en las neuronas GnRH) por la kisspeptina es necesaria para la secreción de GnRH.
La GnRH hipotalámica, a su vez, estimula la secreción de hormonas pituitarias: la hormona foliculoestimulante (FSH) y la hormona luteinizante (LH). La FSH estimula las células de Sertoli en el hombre y produce espermatogonias, y estimula las células foliculares en la mujer, lo que produce la ovulación. La LH regula la producción de testosterona y estrógeno.
El desarrollo puberal anormal puede ser por:
Hipogonadismo hipogonadotrópico que resulta de una falta de producción o acción de gonadotropinas séricas y se manifiesta como una ausencia de desarrollo puberal espontáneo. Esto se presenta en pacientes con trastornos hipotalámico-hipofisarios y en aquellos con un retraso funcional (retraso constitucional, enfermedad crónica subyacente, desnutrición, exceso de ejercicio). El hipogonadismo hipogonadotrófico permanente representa aproximadamente el 10% de los casos de retraso puberal en niños y el 20% en niñas, mientras que el hipogonadismo hipogonadotrófico funcional representa entre el 15% y el 20% de los casos en niños y entre el 20% y el 30% en niñas.[17][18][32]
Hipogonadismo hipergonadotrópico que resulta de trastornos gonadales y se manifiesta con un aumento de las concentraciones de gonadotropina sérica en ausencia de signos puberales a la edad adecuada de la pubertad. Representa aproximadamente entre el 5% y el 10% de los casos de retraso puberal en niños y el 25% en niñas.[17][18][32][Figure caption and citation for the preceding image starts]: Ciclo de retroalimentación hipotalámico-hipofisario-gonadal. GnRH, gonadotropinaseradora de gonadotropinas; FSH, hormona foliculoestimulante; LH, hormona luteinizanteDe la colección del Dr. A. Mehta [Citation ends].
Por término medio, la pubertad tarda de 2 a 5 años en completarse y proporciona un potencial de crecimiento de 25 cm en las niñas y 30 cm en los niños. Las hormonas sexuales estimulan directamente el cartílago de crecimiento, lo que se traduce en un aumento de la velocidad de crecimiento. También se observa un aumento en la secreción de la hormona del crecimiento (GH).[33] El estrógeno ya sea del ovario o aromatizado a partir de la testosterona testicular es el factor que media la mayor respuesta a la GH durante la pubertad.[34]
Clasificación
Clasificación etiológica
Hipogonadismo hipogonadotrópico
Es el resultado de una falta de producción o de la acción de las gonadotropinas séricas, que se manifiesta como una ausencia del desarrollo puberal espontáneo. Suele deberse a una anomalía hipotálamo-hipofisaria. También existen formas parciales que pueden provocar un retraso o detención de la pubertad.
El retraso constitucional y otras causas funcionales de retraso, como las enfermedades crónicas, la malnutrición y el ejercicio excesivo, también provocan concentraciones bajas de gonadotrofinas. Estas causan un retraso temporal o un desarrollo lento de la pubertad.
Hipogonadismo hipergonadotrópico
El aumento de las concentraciones de gonadotropina sérica en ausencia de signos puberales a la edad adecuada para la pubertad sugiere insuficiencia gonadal.
Entre los ejemplos se incluyen los síndromes de Turner y Klinefelter.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad