Investigações
Primeiras investigações a serem solicitadas
ressonância nuclear magnética (RNM) cranioencefálica
Exame
A RNM deve ser solicitada assim que surgir suspeita de demência rapidamente progressiva e deve incluir sequências de T1, T2, imagem ponderada por difusão (IPD), mapa de coeficiente de difusão atenuada (CDA) e recuperação da inversão atenuada por fluidos (FLAIR) na RNM cranioencefálica.[76] Se possível, as sequências IPD e CDA devem ser obtidas nos planos coronal e axial para minimizar o artefato da interface ar-cérebro.[Figure caption and citation for the preceding image starts]: Alterações nos gânglios da base observados na doença de Creutzfeldt-Jakob. (A) Ressonância nuclear magnética (RNM) com recuperação da inversão atenuada por fluidos e (B) RNM com imagem ponderada por difusão do mesmo paciente demonstrando hiperintensidades dos gânglios da base bilaterais (setas). Também há hiperintensidade talâmica, pulvinar e medial, bilateral leveDo acervo pessoal de Dr M. Geschwind [Citation ends].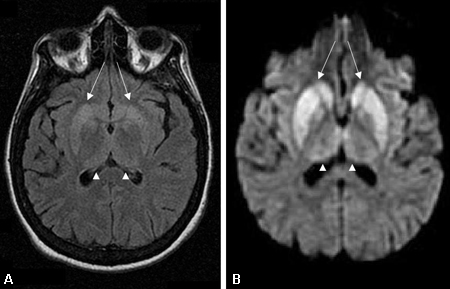 [Figure caption and citation for the preceding image starts]: Hiperintensidade pulvinar e talâmica medial bilateral (setas) em (A) ressonância nuclear magnética (RNM) com recuperação da inversão atenuada por fluidos e (B) RNM com imagem ponderada por difusão em um paciente com doença de Creutzfeldt-Jakob. Esse paciente também apresenta hiperintensidade significativa dos gânglios da base em ambas as sequências (setas)Do acervo pessoal de Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Hiperintensidade pulvinar e talâmica medial bilateral (setas) em (A) ressonância nuclear magnética (RNM) com recuperação da inversão atenuada por fluidos e (B) RNM com imagem ponderada por difusão em um paciente com doença de Creutzfeldt-Jakob. Esse paciente também apresenta hiperintensidade significativa dos gânglios da base em ambas as sequências (setas)Do acervo pessoal de Dr M. Geschwind [Citation ends]. [Figure caption and citation for the preceding image starts]: Manto cortical difuso (setas) observado na (A) ressonância nuclear magnética (RNM) com imagem ponderada por difusão (IPD) e menos proeminente na (B) RNM com recuperação da inversão atenuada por fluidos (FLAIR). Ambas as sequências mostram hiperintensidades do giro do córtex cerebralDo acervo pessoal de Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Manto cortical difuso (setas) observado na (A) ressonância nuclear magnética (RNM) com imagem ponderada por difusão (IPD) e menos proeminente na (B) RNM com recuperação da inversão atenuada por fluidos (FLAIR). Ambas as sequências mostram hiperintensidades do giro do córtex cerebralDo acervo pessoal de Dr M. Geschwind [Citation ends].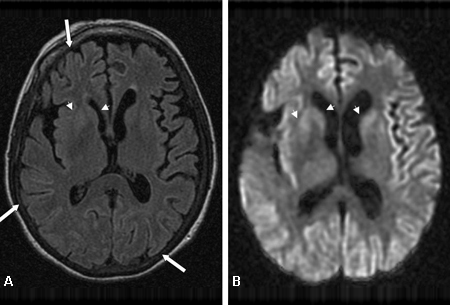
Em geral, as anormalidades encontradas nas sequências de RNM CDA e IPD na doença de Creutzfeldt-Jakob esporádica (eDCJ) e na variante da doença de Creutzfeldt-Jacob (vDCJ) não têm sido relatadas em outras demências semelhantes e são fortemente sugestivas de doença de Creutzfeldt-Jacob (DCJ).[67]
Se o equipamento for de boa qualidade, as varreduras de RNM, IPD e FLAIR podem ter sensibilidade de cerca de 92% a 96% e especificidade de 93% a 94%.[65][67][77]
Nas imagens T1 com gadolínio, a DCJ geralmente não mostra captação de contraste ou anormalidades de intensidade da substância branca; se elas forem encontradas, outros diagnósticos devem ser investigados.[78][79]
Hipointensidade T1 foi relatada no globo pálido na DCJ.[87] O sinal pulvinar, um termo que se refere à hiperintensidade pulvinar bilateral, pode ser observado na vDCJ. Quando o tálamo medial e o pulvinar mostrarem intensidade de sinal, deve-se suspeitar de vDCJ.[71][80][81] Certas formas de DCJ familiar e outras doenças do príon genéticas (particularmente as mutações) apresentam achados de RNM similares àqueles observados na RNM com IPD na eDCJ, mas muitas doenças do príon genéticas mostram apenas atrofia.
Resultado
geralmente demonstra hiperintensidade no córtex cerebral (manto cortical), nos gânglios da base (caudado e putâmen) e no tálamo nas sequências de imagem ponderada por difusão (IPD) e de recuperação da inversão atenuada por fluidos (FLAIR), e hipointensidade (difusão restrita) nas sequências de mapa do coeficiente de difusão atenuado (CDA)
eletroencefalograma (EEG)
Exame
As anormalidades, embora moderadamente específicas, apresentam apenas 60% de sensibilidade e podem não aparecer até os estágios tardios da doença.[14][82]
Quando outras doenças com anormalidades no EEG semelhantes tiverem sido descartadas, esses achados podem ter uma alta especificidade para a doença do príon.[71]
Resultado
lentidão generalizada, focal ou difusa, e complexos periódicos de ondas agudas
Investigações a serem consideradas
conversão induzida por estremecimento (CIE)
Exame
Exame sensível e específico que detecta a isoforma associada à doença da proteína príon no líquido cefalorraquidiano (LCR) em pacientes com doença de Creutzfeldt-Jakob (DCJ) esporádica.[68] QuIC em tempo real (RT-QuIC) e QuIC em ponto final (EP-QuIC) são as versões usadas atualmente por laboratórios diagnósticos, dependendo do país.
Resultado
positiva
biomarcadores do líquido cefalorraquidiano (LCR)
Exame
Embora úteis na confirmação da deterioração neuronal rápida, esses biomarcadores do LCR não podem diagnosticar ou descartar em definitivo a doença do príon.[83][88][89][90] Esses testes devem ser interpretados com cuidado, já que sua sensibilidade e especificidade para a eDCJ ainda não estão claras.
O peptídeo beta-amiloide 42 pode estar diminuído e a tau total (tau-T) e a tau fosforilada (tau-P) podem estar elevadas na doença de Alzheimer.
Relatou-se que a proteína 14-3-3 encontrada no LCR é uma forte indicadora de DCJ. Entretanto, a sensibilidade e a especificidade desse teste variam grandemente na literatura.[71][91][92][93] As diretrizes atuais estão abandonando o uso desse teste.
Alguns estudos sugeriram que as proteínas do LCR, como a 14-3-3, a tau total e a enolase neurônio-específica, possuem sensibilidade mais alta nas doenças rapidamente progressivas, dando suporte à ideia de que elas sejam liberadas durante a lesão e a morte neuronal e não sejam necessariamente específicas para a doença do príon.[71][84][88][89]
Resultado
proteína 14-3-3 (ou seja, western blot) pode ser positiva; proteína tau total (tau-T) elevada (>1200 picogramas/mL); enolase neurônio-específica elevada (>35 nanogramas/mL)
teste genético para gene da proteína príon
Exame
O início e as manifestações clínicas de todas as formas da doença do príon são fortemente influenciados pelo códon polimórfico 129 do gene da proteína príon endógeno (PRNP). O códon 129 pode ser a metionina (M) ou a valina (V). Combinações homozigóticas (por exemplo, MM ou VV) resultam em risco elevado para desenvolvimento da doença do príon. O tipo de príon (tipo 1 ou 2) também influencia a manifestação da doença; entretanto, o tipo de príon só pode ser determinado através de tecido cerebral congelado obtido de biópsias ou de autópsias.[14][23]
As doenças do príon genéticas são causadas por uma mutação no gene que codifica a PRNP, localizada no cromossomo 20.[6] Até o momento, foram identificadas mais de 40 mutações diferentes, cada uma apresentando o seu próprio fenótipo da doença (ou seja, Gerstmann-Straussler-Scheinker, insônia familiar fatal e doença de Creutzfeldt-Jacob [DCJ] familiar).
As mutações PRNP são transmitidas de forma autossômica dominante.[6] É importante ressaltar que cerca de 60% dos pacientes com doença de príon apontada como genética não apresentavam história familiar positiva conhecida da doença de príon. Frequentemente, investigações adicionais revelam uma história familiar de doença de Alzheimer ou Parkinson provavelmente diagnosticada de maneira errônea, ou um genitor que faleceu antes do início dos sintomas.[21][22]
Os pacientes e as famílias devem receber aconselhamento genético e entender as implicações envolvidas antes de conhecerem tais resultados. Como a doença do príon genética é autossômica dominante, o protocolo usado para o teste da doença de Huntington é geralmente seguido.[86] Esse protocolo é usado para várias doenças neurológicas autossômicas dominantes para assegurar que os pacientes, suas famílias e outros entendam as implicações psicológicas, psiquiátricas, clínicas, legais e outras do teste genético.
As amostras devem ser acompanhadas de uma história clínica breve e enviadas no mesmo dia em que foram coletadas.
The National Creutzfeldt-Jakob Disease Research and Surveillance Unit Opens in new window Public Health Agency of Canada: prion disease information Opens in new window National Prion Disease Pathology Surveillance Center Opens in new window
Resultado
positiva
biópsia (cérebro, amígdala)
Exame
Biópsia do cérebro é a única maneira definitiva de diagnosticar a doença do príon antemortem esporádica. Na variante da DCJ, uma biópsia da amígdala pode ser diagnóstica.
Devido ao padrão imprevisível do acúmulo de proteínas no cérebro, um resultado falso-negativo é possível.
O procedimento de biópsia do cérebro pode colocar os pacientes em risco desnecessário de infecção ou dano cerebral adicional. Ainda que o diagnóstico seja confirmado, não há tratamento disponível.
Proteínas do príon são resistentes aos métodos padrão de esterilização cirúrgica, e a equipe médica que realiza o procedimento pode ser colocada em risco de transmissão da DCJ. É necessário seguir protocolos adequados para minimizar a transmissão.[27]
Considerando todos os aspectos, não se recomenda a biópsia do cérebro para diagnosticar a DCJ, especialmente porque a história clínica, a RNM e a QuIC são, atualmente, ferramentas muito úteis para o diagnóstico.[67][71]
Se uma biópsia do cérebro for realizada, o tecido deve ser retirado de áreas de anormalidade mostradas na RNM quando possível.
Embora as marcas microscópicas sejam características dessa doença, outros quadros clínicos também podem apresentar essas alterações.
A presença do PrPSc na imuno-histoquímica ou no western blot é requerida para o diagnóstico definitivo.[3][36]
Encontram-se placas amiloides de PrPSc em aproximadamente 10% dos casos de DCJ, e em casos de Gerstmann-Straussler-Scheinker, um núcleo de PrPSc amiloide é circundado por outro grupo de glóbulos amiloides menores.
A vDCJ também apresenta uma característica patológica relativamente única de um núcleo de placas amiloides de PrPSc circundado por vacúolos. Elas são chamadas de placas floridas, já que se parecem com uma flor.
Na vDCJ, os PrPSc também podem ser identificados no sistema linforreticular durante a evolução da doença, e biópsias da amígdala podem mostrar a presença de PrPSc na vDCJ, embora estejam ausentes em outras formas da doença do príon em humanos.[3][37]
Resultado
microscópica: vacuolização (alterações espongiformes), perda neuronal e astrogliose; histológica: presença de príon patogênico (PrPSc) através de imuno-histoquímica ou western blot; pode apresentar amiloide
O uso deste conteúdo está sujeito ao nosso aviso legal