Abordagem
A apresentação do paciente pode ser bastante útil para se determinar se os sintomas são consistentes com a doença de Creutzfeldt-Jakob (DCJ) ou se podem ser atribuídos a outra condição. Uma abordagem excludente deve ser usada para diagnosticar os pacientes, embora os achados da ressonância nuclear magnética (RNM) com imagem ponderada por difusão (IPD) e o mapa de coeficiente de difusão atenuado (CDA) tenham alta utilidade diagnóstica.[65][66][67] A conversão induzida por estremecimento em tempo real (RT-QuIC) do líquido cefalorraquidiano (LCR) também apresenta alto valor diagnóstico.[68][69][70]
História
O início da DCJ esporádica (eDCJ) é subagudo, mas o progresso geral é bastante rápido em relação às outras demências de desenvolvimento lento, como a doença de Alzheimer. Os potenciais fatores de risco devem ser observados, incluindo certos procedimentos clínicos a que o paciente possa ter sido submetido (DCJ iatrogênica) e detalhes de viagens ao exterior (datas, duração, consumo de carne bovina), particularmente ao Reino Unido, para a variante da DCJ (vDCJ).
Mais de 60% dos pacientes com DCJ portadores de uma mutação no gene da proteína príon (PRNP) não apresentavam história familiar conhecida da doença do príon. É importante obter uma árvore genealógica detalhada, mas é recomendável que todos os pacientes sejam testados para mutações genéticas, mesmo na ausência de história familiar. Deve-se avaliar a história familiar quanto à demência, outras doenças neurológicas e transtornos psiquiátricos, já que as formas genéticas da doença do príon são frequentemente diagnosticadas erroneamente como outras condições.
Exame físico e neurológico
O quadro clínico da doença do príon provavelmente depende das regiões do cérebro nas quais o príon está se acumulando. Os sintomas podem mimetizar outras doenças neurológicas e psiquiátricas. Pacientes com suspeita de doença do príon devem ser encaminhados imediatamente a um neurologista. Um exame físico neurológico detalhado é vital. As características encontradas no exame físico ajudarão a guiar a investigação excludente.
Mais comumente, a eDCJ ocorre na sétima década de vida (paciente na faixa dos 60 anos), e geralmente manifesta-se da seguinte forma:
Queixas cognitivas, falta de coordenação, alterações comportamentais e/ou visuais.[71]
Em um estudo com mais de 100 casos de DCJ, os problemas cognitivos foram os sintomas iniciais mais comuns.[71] Frequentemente, os pacientes vivenciam perda da memória, afasia e dificuldade de executar funções (por exemplo, organizar, planejar e realizar multitarefas).
Sintomas comportamentais, cerebelares e outros sintomas motores foram as manifestações subsequentes mais comuns. As características motoras incluem parkinsonismo, mioclonia e ataxia de marcha e/ou dos membros. O comprometimento dos lobos frontais ou das conexões frontais subcorticais podem afetar o comportamento, causando agitação, depressão e outras características psiquiátricas.[72]
Alguns pacientes também podem descrever sintomas constitucionais ou inespecíficos como vertigem, cefaleia e tontura, que podem preceder a doença por semanas ou até mesmo meses.[73]
Geralmente, os sintomas visuais são menos comuns, mas podem incluir diplopia, alucinações e outras distorções visuais.[72]
A vDCJ manifesta-se de maneira bastante diferente da forma esporádica.
Ela tipicamente afeta adultos jovens e adolescentes.
Na maioria dos pacientes os primeiros sintomas são psiquiátricos, incluindo depressão profunda e comprometimento cognitivo leve.
Mais tarde em sua evolução, os pacientes desenvolvem demência, ataxia, sintomas sensoriais dolorosos e/ou um distúrbio do movimento.[11][21][71]
A DCJ genética ocorre devido a mais de 40 mutações diferentes no gene PRNP e pode ainda ser subdividida em DCJ familiar, Gerstmann-Straussler-Scheinker (GSS) e insônia familiar fatal (IFF) com base nos achados clínicos e patológicos.
A DCJ familiar pode ter uma evolução mais lenta e longa que a GSS ou a IFF.
A evolução clínica da GSS é tipicamente mais lenta e longa que a da eDCJ - frequentemente de alguns anos e de até uma década. Os sinais de apresentação da GSS podem constituir-se de parkinsonismo ou ataxia, que podem ser erroneamente diagnosticados como outra doença neurodegenerativa mais lenta como demências parkinsonianas atípicas, doença de Parkinson e atrofia de múltiplos sistemas.[74]
Geralmente, a IFF manifesta-se como uma síndrome de insônia e de disautonomia. Ataxia ou falta de coordenação cerebelar podem ocorrer. A demência ocorre posteriormente na evolução da doença.[75]
RNM
A RNM deve ser solicitada assim que houver suspeita de uma demência rapidamente progressiva. Os achados são especialmente valiosos no diagnóstico da doença do príon, e têm uma alta sensibilidade e especificidade quando se usa a recuperação da inversão atenuada por fluidos (FLAIR), e particularmente, a imagem ponderada por difusão (IPD) ou o mapa de CDA..[66][67][76][Figure caption and citation for the preceding image starts]: Alterações nos gânglios da base observados na doença de Creutzfeldt-Jakob. (A) Ressonância nuclear magnética (RNM) com recuperação da inversão atenuada por fluidos e (B) RNM com imagem ponderada por difusão do mesmo paciente demonstrando hiperintensidades dos gânglios da base bilaterais (setas). Também há hiperintensidade talâmica, pulvinar e medial, bilateral leveDo acervo pessoal de Dr M. Geschwind [Citation ends].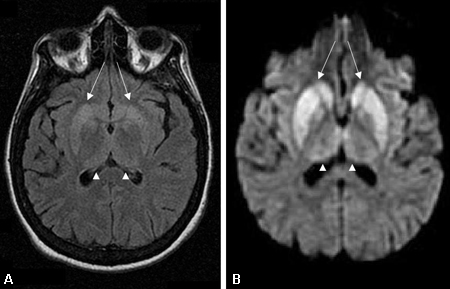 [Figure caption and citation for the preceding image starts]: Hiperintensidade pulvinar e talâmica medial bilateral (setas) em (A) ressonância nuclear magnética (RNM) com recuperação da inversão atenuada por fluidos e (B) RNM com imagem ponderada por difusão em um paciente com doença de Creutzfeldt-Jakob. Esse paciente também apresenta hiperintensidade significativa dos gânglios da base em ambas as sequências (setas)Do acervo pessoal de Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Hiperintensidade pulvinar e talâmica medial bilateral (setas) em (A) ressonância nuclear magnética (RNM) com recuperação da inversão atenuada por fluidos e (B) RNM com imagem ponderada por difusão em um paciente com doença de Creutzfeldt-Jakob. Esse paciente também apresenta hiperintensidade significativa dos gânglios da base em ambas as sequências (setas)Do acervo pessoal de Dr M. Geschwind [Citation ends]. [Figure caption and citation for the preceding image starts]: Manto cortical difuso (setas) observado na (A) ressonância nuclear magnética (RNM) com imagem ponderada por difusão (IPD) e menos proeminente na (B) RNM com recuperação da inversão atenuada por fluidos (FLAIR). Ambas as sequências mostram hiperintensidades do giro do córtex cerebralDo acervo pessoal de Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Manto cortical difuso (setas) observado na (A) ressonância nuclear magnética (RNM) com imagem ponderada por difusão (IPD) e menos proeminente na (B) RNM com recuperação da inversão atenuada por fluidos (FLAIR). Ambas as sequências mostram hiperintensidades do giro do córtex cerebralDo acervo pessoal de Dr M. Geschwind [Citation ends].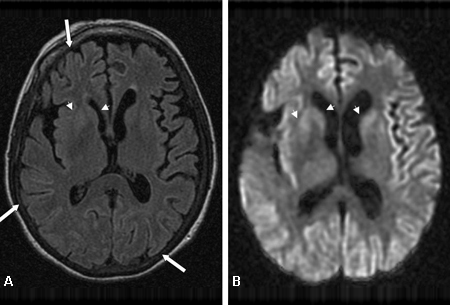
A RNM com FLAIR e IPD demonstram hiperintensidade nos giros da substância cinzenta do córtex cerebral, nos gânglios da base (caudado e putâmen) e, menos comumente, no tálamo.[67][77]
Nas imagens T1 com gadolínio, a DCJ geralmente não mostra captação de contraste ou anormalidades de intensidade da substância branca. Se elas forem encontradas, outros diagnósticos devem ser investigados.[78][79] Alguns pacientes com DCJ apresentam hiperintensidade T1 no globo pálido.
O sinal pulvinar, um termo que se refere à hiperintensidade pulvinar bilateral, pode ser observado na vDCJ, e quando o tálamo medial e o pulvinar mostram intensidade de sinal, deve-se suspeitar de vDCJ.[71][80][81]
Geralmente, as anormalidades de CDA e de IPD encontradas na RNM na eDCJ e na vDCJ não têm sido relatadas em outras demências semelhantes e são fortemente sugestivas de DCJ.[67]
eletroencefalograma (EEG)
O EEG é um exame de rotina. Os achados podem incluir lentidão generalizada, focal ou difusa, e complexos periódicos de ondas agudas. Essas anormalidades, embora moderadamente específicas, apresentam apenas 60% de sensibilidade e podem não aparecer até os estágios tardios da doença.[77][82] Quando outras doenças com anormalidades no EEG semelhantes tiverem sido descartadas, esses achados podem apresentar alta especificidade para a doença do príon.[71]
Teste de LCR
A conversão induzida por estremecimento em tempo real (RT-QuIC) está sendo usada em vários países como ensaio direto para detectar a presença de príons anormais no LCR. Ela apresenta especificidade e sensibilidade altas para DCJ, especialmente as formas esporádicas.[68][69][70]
Relatou-se que a proteína 14-3-3 encontrada no LCR é uma forte indicadora de DCJ; entretanto, a sensibilidade e a especificidade desse teste variam consideravelmente na literatura.[71] Embora haja uma grande discordância sobre a sensibilidade desse teste para a eDCJ, a comunidade neurológica aceita cada vez mais o fato de que ele não seja suficientemente específico para a eDCJ ou para outras doenças priônicas humanas.[83] As diretrizes atuais estão abandonando o uso desse teste.
Estudos de investigação sugeriram que as proteínas do LCR, como tau total (tau-T) e enolase neurônio-específica (ENE), possuem uma sensibilidade equivalente ou ligeiramente superior, mas especificidade muito maior que a 14-3-3. Entretanto, essas proteínas também podem ser elevadas em outras doenças não príon rapidamente progressivas, dando suporte à ideia de que elas são liberadas durante a lesão e a morte neuronal e não são necessariamente específicas para a doença do príon.[84]
Embora úteis na confirmação da deterioração neuronal rápida, esses biomarcadores não podem diagnosticar ou descartar em definitivo a doença do príon.[66][71][83][85]
Exame de sangue e teste genético
Nenhum exame de sangue aprovado para detectar príons está disponível, mas os pacientes devem ser rastreados para mutações genéticas no gene PRNP.
As amostras deverão ser enviadas a um laboratório especializado.
The National Creutzfeldt-Jakob Disease Research and Surveillance Unit Opens in new window
Public Health Agency of Canada: prion disease information Opens in new window
National Prion Disease Pathology Surveillance Center Opens in new window
Recomenda-se que os pacientes e as famílias recebam aconselhamento genético e entendam as implicações antes de serem submetidos aos testes e de conhecerem os resultados genéticos. O protocolo de aconselhamento genético usado para o teste da doença de Huntington é geralmente seguido para PRNP.[86]
Biópsia
Biópsia do cérebro é a única maneira definitiva de diagnosticar a doença do príon antemortem esporádica. Na variante da DCJ, uma biópsia da amígdala pode ser diagnóstica.
Devido ao padrão imprevisível do acúmulo de proteínas no cérebro, é possível que a biópsia indique um resultado falso-negativo. O procedimento pode colocar os pacientes em risco desnecessário de infecção ou dano cerebral adicional. Mesmo que o diagnóstico seja confirmado, ainda não há tratamento disponível.
Proteínas do príon também são resistentes aos métodos padrão de esterilização cirúrgica, e a equipe médica que realiza o procedimento pode ser colocada em risco de transmissão da DCJ. Estão disponíveis diretrizes do UK Department of Health para minimizar o risco de transmissão.[27] A biópsia do cérebro é recomendada apenas quando a RNM for negativa para DCJ e todas as outras doenças foram excluídas com outros métodos menos invasivos.[71]
Autópsia
A autópsia é fortemente estimulada, já que a confirmação patológica é a única maneira definitiva de diagnosticar a doença do príon fora da biópsia.[36]
O uso deste conteúdo está sujeito ao nosso aviso legal