Etiologia
O pseudo-hipoparatireoidismo (PHP) ocorre como resultado de mutações na cascata de sinalização a jusante do paratormônio (PTH)/receptor do peptídeo relacionado ao PTH (PTHR1). Todas as mutações identificadas até hoje afetam o GNAS, o gene codificante da proteína de ligação ao nucleotídeo guanina (proteína G), a Gs-alfa.[11][12][13] No entanto, não é conhecida a mutação causadora de todas as formas de PHP. A transcrição do gene da Gs-alfa (posicionado no cromossomo 20q13.3 no locus GNAS) é afetada pelo imprinting genômico, um fenômeno epigenético (não determinado por sequência de ácido desoxirribonucleico [DNA]) que causa a expressão aberrante ou inadequada da Gs-alfa. O "imprinting" de um gene permite que uma célula expresse somente um alelo, mais comumente de origem materna, mas a herança paterna também foi descrita. Esses efeitos metabólicos de origem parental são decorrentes da expressão preferencial do alelo materno em um pequeno número de tecidos.[14] O silenciamento ou metilação ineficazes dos genes GNAS também podem resultar nos achados de PHP. Não são completamente conhecidos quais tecidos estão envolvidos nos efeitos metabólicos de origem parental da mutação na Gs-alfa. O pequeno número de tecidos afetados ajuda a explicar a apresentação única desses pacientes. Foi sugerido que a etiologia da obesidade é resultante do gasto reduzido de energia de uma mutação na Gs-alfa que sofreu imprinting no núcleo paraventricular do hipotálamo.[15] O tipo de PHP depende da mutação GNAS, do tecido que sofreu imprinting e do padrão de herança.
O tipo 1a é causado por perda de função devido a mutação heterozigótica na Gs-alfa herdado do lado materno em seres humanos e outros mamíferos.[16][17][18][19][20] Essa redução na sinalização acoplada à proteína G pode resultar na falta da função do PTH, hormônio estimulante da tireoide (TSH), gonadotrofinas e hormônio de liberação do hormônio do crescimento. Além disso, resulta um fenótipo esquelético característico, incluindo baixa estatura, compleição atarracada, obesidade, rosto redondo, hipoplasia dental, braquimetacarpia, braquimetatarsia e calcificação/ossificação de tecido mole. É relatado como osteodistrofia hereditária de Albright.
O tipo 1b é causado por mutações esporádicas ou autossômicas dominantes menos graves no locus GNAS, que resultam em redução da expressão da Gs-alfa, afetando os tecidos sensíveis ao PTH (principalmente, os rins) e, às vezes, os tecidos sensíveis ao TSH (a glândula tireoide). Outros tecidos são preservados e, portanto, não há anormalidades morfológicas.
O tipo 1c é normalmente associado à atividade normal da Gs-alfa e à ausência de mutações GNAS. No entanto, mutações na carboxila terminal do GNAS têm sido detectadas em um subgrupo de pacientes do tipo 1c.[7] O fenótipo do tipo 1c é semelhante ao do tipo 1a, com resistência hormonal generalizada e osteodistrofia hereditária de Albright. A resposta de adenosina monofosfato cíclica (cAMP) é atenuada no tipo 1c, sugerindo que há um defeito na síntese de cAMP e ruptura do ciclo.
O tipo 2 é causado por mutações de perda de função ainda não identificadas sem mutação nos exons codificadores da Gs-alfa. O fenótipo do tipo 2 é mais leve, com morfologia normal e resistência seletiva ao PTH. A resposta de cAMP ao PTH é preservada no pseudo-hipoparatireoidismo tipo 2, sugerindo que o defeito está localizado a jusante do cAMP.[21]
O pseudopseudo-hipoparatireoidismo é causado pelas mesmas mutações que o PHP tipos 1a e 1b, mas as mutações são hereditárias do pai e não da mãe. A herança paterna não produz resistência ao hormônio, mas o fenótipo esquelético característico ainda ocorre. A razão para isso é que a Gs-alfa é primariamente expressa a partir do alelo materno nos rins. Se o alelo materno tiver uma mutação não funcional, a função mediada pela Gs-alfa será perdida nos tecidos-alvo, causando PHP. No entanto, se o alelo paterno for portador da mutação, a função mediada pela Gs-alfa será preservada nos tecidos-alvo.[22][23][24][25] Como os tecidos afetados pelo fenótipo esquelético característico requerem os dois alelos para que sejam totalmente funcionais, essas anormalidades são observadas em alguns tipos de PHP e em pacientes com pseudo-PHP.
Fisiopatologia
O paratormônio (PTH) está envolvido primariamente na homeostase do cálcio. Ele é liberado pela glândula paratireoide quando um receptor de cálcio percebe um decréscimo no nível de cálcio ionizado. O PTH libera cálcio dos ossos, aumenta a absorção intestinal do cálcio ao promover a síntese e ativação da vitamina D, e promove reabsorção do cálcio e excreção do fosfato nos rins. A ligação do PTH ou do peptídeo relacionado ao PTH (PTHRP), uma proteína importante para o crescimento, ao seu receptor é seguida por uma cascata de eventos que são mediados por proteínas G. Quando o ligante se liga ao seu receptor, a proteína G é ativada e estimula a síntese do segundo mensageiro, a adenosina monofosfato cíclica (cAMP). Defeitos na sinalização de cAMP podem resultar em anormalidades das funções metabólica, esquelética ou mental superior decorrentes da ampla distribuição desse sistema de sinalização. Os defeitos mais comuns observados no pseudo-hipoparatireoidismo são resistência ao PTH (produzindo hipocalcemia), resistência ao hormônio estimulante da tireoide (produzindo hipotireoidismo) e o fenótipo esquelético característico (osteodistrofia hereditária de Albright). A resistência ao PTH resulta em hipocalcemia. Os níveis de PTH se tornam cronicamente elevados porque a hipocalcemia e hiperfosfatemia continuam a estimular a produção do PTH, o qual, por sua vez, não consegue restaurar os níveis de cálcio.
Muitos pacientes apresentam hipocalcemia crônica assintomática, mas podem ocorrer sintomas de hipersensibilidade de nervos e músculos, parestesias, tremores, ansiedade e anormalidades eletrocardiográficas (prolongamento de QT). Se os níveis de cálcio não forem corrigidos nos pacientes com hipocalcemia sintomática, arritmia cardíaca e morte podem ocorrer.
A resistência ao PTH também pode causar hiperfosfatemia, que estimula a secreção do PTH na tentativa de reduzir os níveis de fosfato. A hiperfosfatemia causa a formação aumentada de fosfato de cálcio em locais extraesqueléticos. O fosfato de cálcio pode ser depositado nos rins (causando nefrolitíase), no cérebro (causando calcificação dos gânglios da base), na pele (causando calcificação e ossificação subdérmica) e no olho (causando catarata).
O fenótipo esquelético característico não apresenta relação com a sensibilidade ao PTH. Ele se deve ao papel crucial da sinalização da Gs-alfa no crescimento, diferenciação e estrutura desses tecidos.[Figure caption and citation for the preceding image starts]: Visão geral da regulação do cálcio sérico. As linhas duplas e a linha pontilhada indicam os defeitos na sinalização do paratormônio (PTH) observados no pseudo-hipoparatireoidismo. O defeito na resposta renal é mais pronunciadoCriado no BMJ Evidence Centre com base nas informações do autor [Citation ends].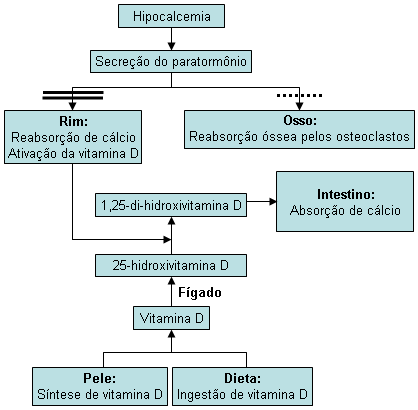 [Figure caption and citation for the preceding image starts]: A cascata de sinalizações do paratormônioDo acervo de Kent Wehmeier, Universidade da Flórida, Jacksonville [Citation ends].
[Figure caption and citation for the preceding image starts]: A cascata de sinalizações do paratormônioDo acervo de Kent Wehmeier, Universidade da Flórida, Jacksonville [Citation ends].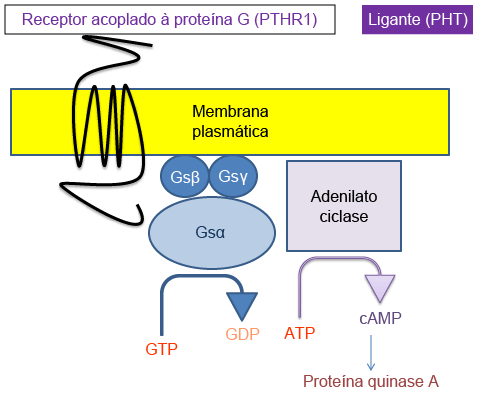
Classificação
Tipos de pseudo-hipoparatireoidismo (PHP) com base nas características clínicas, resposta ao paratormônio (PTH) e genótipo[3][4][5]
Tipo 1a:
Causado por perda de função devido a mutação heterozigótica acima ou dentro do locus do GNAS, onde o gene de 13 exons codifica a subunidade alfa da proteína estimuladora de ligação ao nucleotídeo guanina (proteína G) Gs-alfa
Osteodistrofia hereditária de Albright associada
Resposta reduzida ao PTH exógeno, conforme medida pela adenosina monofosfato cíclica (cAMP) na urina e fósforo na urina
O cálcio sérico é baixo
A resistência ao hormônio é generalizada, afetando hormônios que precisam da sinalização da Gs-alfa, como o PTH/peptídeo relacionado ao PTH (PTHRP), hormônio estimulante da tireoide (TSH), gonadotrofinas e hormônio de liberação do hormônio do crescimento
Herança autossômica dominante; ligada ao alelo materno.
Tipo 1b:
Causado por mutações no GNAS resultantes da ruptura dos elementos de controle de "imprinting"
Transcrição reduzida de Gs-alfa em determinados tecidos onde a transcrição do gene é derivada do alelo materno
Nenhuma alteração significativa na atividade da Gs-alfa é detectada
A resistência hormonal é limitada à atividade do PTH no córtex renal
Não há fenótipo esquelético associado
Resposta reduzida ao PTH exógeno, conforme medida pelo cAMP na urina e fósforo na urina
O cálcio sérico é baixo
A resistência hormonal é limitada ao tecido-alvo do PTH; no entanto, há números crescentes de casos associados à resistência ao TSH
A herança pode ser esporádica ou autossômica dominante familiar.
Tipo 1c:
A atividade normal da Gs-alfa com a ausência de mutações no GNAS são as características do tipo 1c.[6] No entanto, duas mutações nonsense e duas mutações de sentido incorreto na extremidade carboxila do GNAS foram detectadas em alguns tipos de pacientes do tipo 1c[6][7][8]
Osteodistrofia hereditária de Albright associada
Resposta reduzida ao PTH exógeno, conforme medida pelo cAMP na urina e fósforo na urina
O cálcio sérico é baixo
A resistência hormonal é generalizada (afeta qualquer hormônio que depende da sinalização da Gs-alfa)
Herança semelhante à do tipo 1a (ou seja, autossômica dominante, ligada ao alelo materno).
Tipo 2:
A mutação genética subjacente é desconhecida
Não há fenótipo esquelético associado
Resposta complexa ao PTH exógeno, a resposta do fósforo na urina é reduzida, mas a do cAMP na urina é normal
O cálcio sérico é baixo
A resistência hormonal é limitada ao tecido-alvo do PTH
A herança é desconhecida.
Pseudopseudo-hipoparatireoidismo:
Causado por mutações no GNAS
Osteodistrofia hereditária de Albright associada
Resposta normal ao PTH exógeno, conforme medida pelo cAMP na urina e fósforo na urina
O cálcio sérico é normal
Sem resistência hormonal
Herança autossômica dominante, ligada ao alelo paterno.
O uso deste conteúdo está sujeito ao nosso aviso legal