Abordagem
A maioria dos pacientes que apresentam um retardo funcional ou temporário da puberdade progride espontaneamente e atinge um potencial de crescimento adequado. No entanto, os pacientes que apresentam um problema orgânico, como os indivíduos com deficiência congênita de gonadotropina ou anormalidades gonadais, precisam de tratamento precoce para atingir a altura final ideal e as características sexuais secundárias. Na ausência de alterações puberais, a velocidade da altura cai de 5 cm por ano aos 12 anos de idade em 1 cm ao ano para cada ano de ausência de manifestação da puberdade. Os pacientes devem ser avaliados por um endocrinologista pediátrico com experiência no diagnóstico e no tratamento da puberdade tardia.
História
A maioria dos pacientes que apresentam puberdade tardia é composta por meninos. Muitos pacientes procuram ajuda médica devido ao crescimento lento em vez do desenvolvimento puberal lento, bem como devido a preocupações de que a puberdade ainda não tenha começado.
Inicialmente, a avaliação deve incluir uma história detalhada para causas de um atraso temporário. Quaisquer afecções clínicas crônicas podem causar puberdade tardia, com ou sem baixa estatura, e pouco ganho de peso. As doenças comuns incluem desnutrição, cardiopatia crônica, asma moderada a grave, fibrose cística, doença celíaca, doença inflamatória intestinal (doença de Crohn e colite ulcerativa), distúrbios inflamatórios (por exemplo, artrite idiopática juvenil), insuficiência renal crônica, qualquer neoplasia maligna crônica e diabetes mellitus mal controlado. É necessário investigar qualquer história de padrões alimentares anormais ou perda de peso recente em meninas. Esses eventos podem, ocasionalmente, estar associados a rotinas de exercícios intensas e devem levantar suspeita de transtorno alimentar, como anorexia nervosa ou bulimia nervosa.
O atraso constitucional, também conhecido como retardo constitucional do crescimento e da puberdade (RCCP), puberdade tardia autolimitada ou puberdade tardia isolada, é a causa mais comum de puberdade tardia em homens e mulheres.[1][20][37] Ele tende a ocorrer na mesma família e está associado a baixa estatura. Uma história familiar deve ser registrada, incluindo a altura de ambos os pais e suas idades no início do desenvolvimento puberal, bem como a idade da menarca materna.[37]
A história pode revelar cirurgia prévia ou radioterapia no cérebro (resultando em deficiência de gonadotropina), abdome ou pelve (danos gonadais) em decorrência de malignidade prévia. Alguns agentes quimioterápicos podem causar puberdade tardia. Dor testicular ou infecções virais recentes, como caxumba, podem indicar infecção ou torção testicular. Histiocitose, doença falciforme e sobrecarga de ferro (associada à transfusão) podem causar deficiência permanente de gonadotropina.
Pode haver sintomas sugestivos de outros deficits hormonais, como deficit de crescimento decorrente de deficiência de hormônio do crescimento ou sintomas de hipotireoidismo, que podem indicar uma causa central (hipotálamo-hipófise). Esses eventos podem estar associados à perda do olfato (anosmia), a qual sugere síndrome de Kallman.
Várias anormalidades das glândulas endócrinas, como a puberdade tardia associada ao diabetes mellitus e/ou doença de Addison, devem levantar suspeita de uma doença autoimune. Raramente, a deficiência congênita genética de gonadotropina pode estar associada a hipoplasia adrenal congênita, e a história pode indicar perda de sal ou crise adrenal. É provável que o diagnóstico já esteja estabelecido; neste caso, o paciente pode estar recebendo um tratamento com glicocorticoide e mineralocorticoide.[Figure caption and citation for the preceding image starts]: Crescimento em um paciente com retardo constitucional do crescimento e da puberdade; X indica a idade óssea; a seta indica a idade do início do desenvolvimento puberalDo acervo de Dra. A. Mehta [Citation ends].
Estágios de Tanner
O desenvolvimento puberal e sua evolução são avaliados de maneira mais adequada pelos estágios de Tanner.[38][39]
O primeiro sinal de puberdade em meninos é o aumento do tamanho dos testículos. Esse aumento é seguido por alterações penianas e escrotais. O tamanho dos testículos é documentado como a medida do eixo mais longo ou o volume dos testículos medido pelo orquidômetro de Prader.
[Figure caption and citation for the preceding image starts]: Orquidômetro de PraderCriado pelo BMJ Knowledge Centre [Citation ends].

[Figure caption and citation for the preceding image starts]: Método de comparação do tamanho testicular usando o orquidômetro de PraderDo acervo de Dra. A. Mehta [Citation ends].
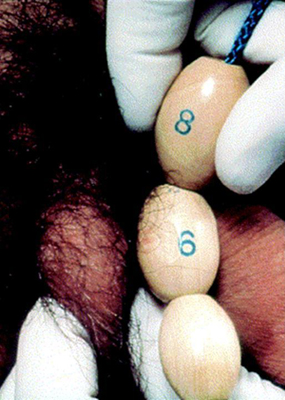 Um volume de 4 mL ou comprimento longitudinal de 2.5 cm define o início da puberdade.[39] O crescimento de pelos axilares e outras mudanças, como alterações da voz e aumento da velocidade de crescimento, ocorrem entre o meio e o final da puberdade. Os pelos faciais não aparecem até a fase final da puberdade.
Um volume de 4 mL ou comprimento longitudinal de 2.5 cm define o início da puberdade.[39] O crescimento de pelos axilares e outras mudanças, como alterações da voz e aumento da velocidade de crescimento, ocorrem entre o meio e o final da puberdade. Os pelos faciais não aparecem até a fase final da puberdade.O primeiro sinal visível de puberdade nas meninas é o desenvolvimento das mamas. Deve-se usar de cautela ao examinar o tecido mamário em meninas obesas, pois a gordura pode ser confundida com tecido mamário. Os pelos púbicos e axilares, a acne e o odor corporal se desenvolvem como resultado de androgênios secretados pela glândula adrenal. O pico do estirão de crescimento ocorre no estágio 3 de desenvolvimento mamário de Tanner, e a menarca tipicamente ocorre em conjunto com o estágio 4 de desenvolvimento mamário de Tanner.[Figure caption and citation for the preceding image starts]: Estágios de Tanner: A, padrões de classificação genital em meninos; B, padrões de classificação de pelos púbicos em meninos; C, padrões de classificação mamária em meninas; D, padrões de classificação de pelos púbicos em meninasAdaptado de Marshall WA, Tanner JM. Arch Dis Child. 1970;45:13-23; Marshall WA, Tanner JM. Arch Dis Child. 1969;44:291-303 [Citation ends].

Exame
É necessário registrar, com precisão, as medidas atuais e prévias de altura e peso. A maioria dos pacientes com puberdade tardia é formada por meninos com retardo constitucional que apresentam um exame normal, com exceção da baixa estatura e da velocidade de crescimento um pouco abaixo da média para a idade e o sexo. Os pacientes com atraso temporário, como os que apresentam doenças crônicas, podem ter as anormalidades típicas desse distúrbio. É importante avaliar o paciente para determinar sintomas de desnutrição.
Os pacientes devem ser avaliados quanto a qualquer desproporção corporal. Os meninos com síndrome de Klinefelter apresentam estatura alta, com um maior crescimento dos membros inferiores. Além disso, eles podem ter testículos disgenéticos (pequenos e macios à palpação) e atraso no desenvolvimento, e podem ter ginecomastia.[27]
Tipicamente, as meninas com síndrome de Turner têm baixa estatura com algumas características dismórficas que incluem linha do cabelo posterior baixa, pescoço alado e orelhas proeminentes com rotação posterior. Em muitos casos, a presença dos sintomas pode ser mais sutil, e é necessário realizar investigações para descartar a síndrome de Turner em qualquer menina que apresente puberdade tardia.[40][41] Consulte Síndrome de Turner.
Pode haver outros sintomas que sugiram um diagnóstico de síndrome (por exemplo, Prader-Willi, Bardet-Biedl, CHARGE [caracterizada por coloboma, atresia das cóanas e canais semicirculares anormais] ou displasia septo-ótica).
É necessário realizar um exame detalhado do sistema nervoso central para procurar quaisquer deficits focais que sugiram tumor intracraniano. O olfato deve ser avaliado; a anosmia sugere síndrome de Kallman, que é uma associação de hipogonadismo hipogonadotrófico orgânico e nervos olfatórios hipoplásicos.[21] Alguns pacientes com síndrome de Kallman apresentam uma falha genética em genes como ANOS1, FGF8, FGFR1, PROK2 ou PROKR2.
[Figure caption and citation for the preceding image starts]: Avaliação de uma paciente com puberdade tardia. RCCP, retardo constitucional do crescimento e desenvolvimento puberal; PT, puberdade tardia; T4L, t4 livre; DGH, deficiência de GH; PRL, prolactina; TFT, teste de função tireoidianaEndocr Rev. 2019 Oct; 40(5): 1285–1317; usado com permissão [Citation ends].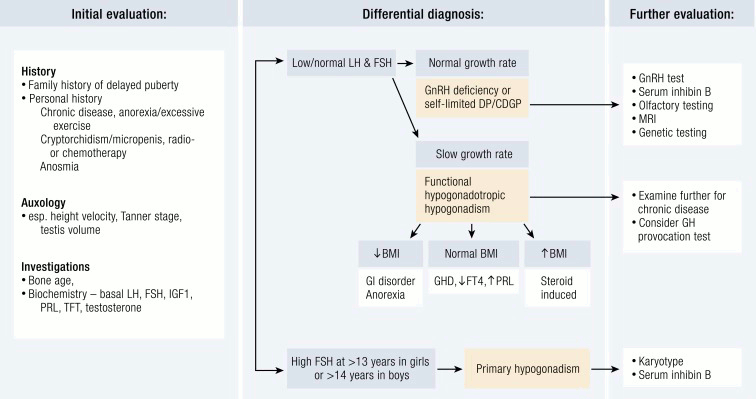
Investigações iniciais
As investigações iniciais devem incluir uma radiografia do punho não dominante (em geral, o esquerdo) para calcular a idade óssea. A idade óssea indica se o atraso constitucional está presente ou ausente e ajuda a predizer a faixa de altura adulta estimada e sua relação com a altura média dos pais.[1] A aparência radiográfica de centros epifisários representativos é comparada aos padrões apropriados publicados para a idade e o sexo. O método mais comumente usado é o de Greulich e Pyle.[42]
A medida laboratorial das concentrações séricas de hormônio folículo-estimulante (FSH) e hormônio luteinizante (LH) ajudará a diferenciar os pacientes com hipogonadismo hipogonadotrófico (níveis baixos ou normais) e com hipogonadismo hipergonadotrófico (níveis elevados).[1] As medições devem ser realizadas em uma amostra de sangue extraída pela manhã, usando um ensaio pediátrico ultrassensível. Como o eixo hipotálamo-hipófise-gonadal permanece quiescente até cerca de 10 a 12 anos de idade, os níveis desses hormônios nas crianças <12 anos devem ser interpretados com cautela. Pode ser difícil distinguir entre o retardo constitucional e a deficiência orgânica de gonadotropina, pois as concentrações basais de gonadotropina podem estar baixas ou normais nos dois grupos.
Investigações e manejo adicionais dependem da história, da gravidade do atraso, da altura e dos resultados da medição de gonadotropina basal.
Exames posteriores
Exame de hormônio liberador de gonadotropina (GnRH)
Pode ser considerado em todos os pacientes com gonadotrofinas basais baixas; no entanto, a sensibilidade e a especificidade são limitadas.[43] As meninas com síndrome de Turner e outros pacientes com anormalidades gonadais, em geral, apresentam elevações pronunciadas nas concentrações séricas basais de FSH e LH (na síndrome de Turner, de 8 a 9 anos de idade), e o teste do GnRH geralmente não é indicado nestes pacientes.[44]
O hormônio liberador de LH (LHRH) é usado para estimular as gonadotrofinas. Ele foi estudado extensivamente em pacientes com gonadotrofinas basais baixas, mas nem sempre ajuda a diferenciar entre o atraso constitucional e a deficiência de gonadotrofina orgânica, pois as concentrações séricas estimuladas de FSH e LH após o LHRH podem ser baixas em ambos os grupos, ou podem estar normais e, portanto, falsamente tranquilizadora em um indivíduo com deficiência hipotalâmica de GnRH, mas com função hipofisária preservada.[45] Deste modo, não fica claro se um indivíduo evoluirá espontaneamente para a puberdade ou terá um problema permanente. Alguns pacientes com um atraso temporário podem, portanto, precisar ser tratados com esteroides sexuais para induzir a puberdade, mas quando reavaliados após a conclusão do crescimento e da puberdade, com um uma tentativa de suspensão do tratamento, podem revelar concentrações séricas de FSH e LH normais.
Inibina B e hormônio antimülleriano
A inibina B basal é uma investigação bioquímica promissora para o diagnóstico da puberdade tardia. Nos homens ela é produzida exclusivamente pelas células de Sertoli dos testículos, e nas mulheres pelas células da granulosa ovariana e da placenta. Uma inibina B baixa de menos de 35 pg/mL em meninos pré-púberes é um marcador sensível para discriminar o hipogonadismo hipogonadotrófico permanente da puberdade tardia constitucional ou autolimitada.[46] Nos homens, o trio de testículos pequenos (limite: 1.1 mL), baixo LH estimulado máximo no teste de estimulação com GnRH (limite: 4.3 UI/L) e baixa concentração basal de inibina B (limite: 61 pg/mL) foram propostos como o discriminador mais efetivo entre essas duas condições.[18] Em homens com hipogonadismo hipergonadotrófico, uma inibina B sérica baixa reflete uma falência primária das células germinativas. A utilidade da inibina B foi menos claramente demonstrada nas meninas.
O hormônio antimülleriano (HAM) é produzido nos homens a partir das células testiculares de Sertoli desde o momento da diferenciação testicular até a puberdade. Nas mulheres, ele é secretado pelas células da granulosa ovariana desde o nascimento até a menopausa, embora as concentrações nas meninas sejam significativamente menores. Pacientes com gônadas disgenéticas têm um HAM sérico baixo. HAM e inibina B indetectáveis são característicos de anorquia congênita, mas também podem ser observados em homens e mulheres com hipogonadismo hipogonadotrófico grave. O HAM e a inibina B podem ser usados, portanto, durante a avaliação da função e da capacidade testicular ou ovariana (como um marcador do potencial para fertilidade) e como ferramenta diagnóstica em conjunto com gonadotrofinas séricas para os hipogonadismos hipo e hipergonadotrófico.[47]
Teste de estímulo com gonadotrofina coriônica humana (hCG)
O exame de hCG é usado para estimular a produção testicular de testosterona, e há indícios de que seja útil em alguns pacientes quando combinado ao exame de LHRH para ajudar a distinguir o retardo constitucional do hipogonadismo hipogonadotrófico.[45]
Observa-se um aumento da concentração sérica de testosterona no retardo constitucional e uma redução da resposta à testosterona no hipogonadismo hipogonadotrófico.
Amostras de gonadotropinas minutadas de repouso (overnight)
Pode demonstrar pulsatilidade, mas sua função no prognóstico de longo prazo do retardo não está clara. É realizado principalmente no âmbito da pesquisa.
Outros testes
Esteroides sexuais: a medição dos esteroides sexuais é útil apenas no contexto dos exames de estimulação por hCG. A medição basal dos esteroides sexuais séricos (testosterona e estradiol) raramente é diagnóstica.
Análise cromossômica: pode revelar síndrome de Klinefelter (47XXY) em meninos, ou síndrome de Turner (45X) em meninas. O diagnóstico da síndrome de Turner (45X) deve ser considerado em todas as meninas de baixa estatura, com ou sem outras características fenotípicas sugestivas da síndrome de Turner.[41] Dependendo do mosaicismo, a disgenesia gonadal mista de 45X/46XY pode se apresentar com genitália ambígua ao nascimento ou com puberdade tardia na adolescência.[40] O microarray cromossômico pode ser indicado em pacientes com hipogonadismo hipergonadotrófico com anormalidades congênitas adicionais ou características sindrômicas.[48]
Sequenciamento genético: para indivíduos com suspeita de hipogonadismo hipogonadotrófico congênito ou síndrome de Kallmann, testes de painel de sequenciamento de nova geração já estão disponíveis no Reino Unido e em algumas outras regiões do mundo.[49]
Imagem: o exame de ultrassonografia pélvica deve ser considerado em meninas com síndrome de Turner ou outras formas de disgenesia gonadal, bem como em meninas com hipogonadismo hipogonadotrófico. Em indivíduos com hipogonadismo hipergonadotrófico, pode revelar gônadas disgenéticas. Naqueles com hipogonadismo hipogonadotrófico, ele é útil para a avaliação da maturidade uterina e ovariana (volume, forma e espessura endometrial) para diagnóstico e antes do início do tratamento para permitir o monitoramento da resposta à terapia. A ultrassonografia abdominal pode revelar anormalidades renais, como agenesia frenal ou rim em ferradura. A ecocardiografia nas meninas com síndrome de Turner pode evidenciar problemas cardíacos, inclusive coarctação da aorta e valva aórtica bicúspide. A imagem de ressonância nuclear magnética do cérebro deve ser considerada se as gonadotrofinas basais estiverem baixas na ausência de história familiar de puberdade tardia. É obrigatório se outros níveis de hormônio hipofisário estiverem deficientes. A neuroimagem do cérebro ajuda a identificar anormalidades estruturais do eixo hipotálamo-hipófise, problemas na linha média cerebral, hipoplasia olfatória e tumores na hipófise.[1][50]
Em pacientes com hipogonadismo hipogonadotrófico confirmado, é necessário considerar outros testes de hormônio da hipófise para descartar deficiências combinadas da glândula.
Medição de anticorpos ovarianos séricos para identificar um processo autoimune.
Testes de função tireoidiana e concentrações séricas de prolactina são úteis para identificar pacientes com hipotireoidismo ou hiperprolactinemia; os grupos podem apresentar puberdade tardia.
Para avaliar a produção de GnRH pelo hipotálamo, a medição de LH em resposta à estimulação com kisspeptina foi proposta como um teste útil para identificar indivíduos com deficiência de GnRH e, portanto, hipogonadismo hipogonadotrófico permanente. A kisspeptina estimula a pulsatilidade do GnRH, promovendo assim a secreção do LH e, em menor grau, do FSH. Um estudo clínico revelou que o aumento máximo de LH após a administração de kisspeptina foi mais preciso para o diagnóstico de homens com deficiência de GnRH do que o teste de estimulação com GnRH.[51] Em um estudo paralelo em adolescentes com atraso puberal (3 mulheres e 13 homens), o pico de LH após a estimulação com kisspeptina demonstrou ser superior ao teste de estimulação com GnRH para predizer a capacidade de progressão durante a puberdade.[52] Embora mais pesquisas sejam necessárias para delinear os parâmetros do uso da kisspeptina na prática clínica pediátrica, esta é uma área promissora para o diagnóstico bioquímico da deficiência de GnRH na adolescência.
O uso deste conteúdo está sujeito ao nosso aviso legal