Monitoramento
O rastreamento regular deve ser realizado, incluindo:[5]
Testes de função tireoidiana (TFTs) para doença tireoidiana autoimune anualmente; anticorpos antitireoidianos podem ser medidos se os TFTs forem anormais
TFHs para o rastreamento de "hepatite de Turner" após os 10 anos de idade, repetidos anualmente
LH e FSH, repetidos anualmente
Medições da PA para hipertensão em todas as consultas
Glicemia de jejum e HbA1c para risco de diabetes, repetidos anualmente após os 10 anos de idade
Lipídeos séricos para monitorar dislipidemia, repetido anualmente após os 18 anos de idade, se houver pelo menos um fator de risco para doença cardiovascular
Rastreamento para perda auditiva condutiva e/ou neurossensorial, repetido a cada 3 anos na infância e a cada 5 anos na fase adulta[5]
Rastreamento de erros refrativos e anormalidades visuais[5]
Estrabismo, ambliopia, hiperopia e miopia, ptose e cegueira de cores foram relatados. Meninas com síndrome de Turner têm uma dobra epicântica (dobra na pálpebra superior que cobre o canto interno do olho) e hipertelorismo (maior distância entre os olhos). Um exame oftalmológico abrangente deve ser realizado entre 12 e 18 meses de idade, ou no momento do diagnóstico, se ocorrer em idade mais avançada.
Rastreamento para anomalias renais congênitas[5]
Ao diagnóstico, realiza-se uma ultrassonografia renal para avaliar a presença de anomalias estruturais, como rim em ferradura, agenesia renal e sistema coletor duplicado, que afetam aproximadamente 25% das pacientes com síndrome de Turner.[3][4]
A função renal geralmente é normal, mas a obstrução do sistema coletor está associada à infecção urinária e pode requerer correção.
Rastreamento para defeitos cardiovasculares congênitos
A coarctação aórtica e as valvas aórticas bicúspides são as anormalidades mais comuns. Outras anormalidades cardíacas incluem drenagem anômala parcial das veias pulmonares, hipoplasia do coração esquerdo e dilatação aórtica. A prevalência de defeitos cardiovasculares é muito maior naquelas com clara evidência de linfedema fetal, como pescoço alado.
Uma valva bicúspide funcional como resultado da fusão completa ou parcial dos folhetos coronarianos direito e esquerdo é observada em 30% das pacientes assintomáticas e representa um risco de infecção, deterioração valvar e dilatação e dissecção aórtica.[18] Defeitos cardiovasculares congênitos constituem a principal causa de mortalidade prematura na síndrome de Turner. Dois genes, TIMP1 e TIMP3, que estão ambos localizados no braço curto do cromossomo X quando o hemizigoto aumenta o risco de aortopatia com razão de chances de 12.86.[19]
Deve-se tentar visualizar a valva aórtica, a aorta torácica e as veias pulmonares por ecocardiografia transtorácica (ETT) em lactentes ou por ressonância nuclear magnética cardíaca (RNMC) ou TC em crianças mais velhas e adultos. Se nenhuma anomalia for detectada e a PA sistêmica for normal, deve-se realizar uma reavaliação do sistema cardiovascular com ETT ou RNMC a cada 5-10 anos durante a idade adulta, especialmente antes de uma gravidez desejada ou com o desenvolvimento de hipertensão.
A densidade mineral óssea basal deve ser avaliada na transição para o atendimento de adultos e reavaliada a cada 5 anos em mulheres adultas, e ao descontinuar o estrogênio.[5]
O nível de imunoglobulina A (IgA) transglutaminase tecidual (se houver níveis de IgA normais) deve ser medido a cada 2 anos a partir dos 2 anos de idade e, em adultos, quando houver sintomas sugestivos, para monitorar a doença celíaca.[5]
A avaliação cardíaca deve ser realizada com ETT ou RNMC a cada 1-5 anos, dependendo da gravidade, ou conforme indicação médica.[5]
Defeitos cardiovasculares congênitos constituem a principal causa de mortalidade prematura na síndrome de Turner. Todas as meninas até os 12 anos de idade devem realizar um rastreamento cardíaco por ressonância nuclear magnética (RNM), caracterização minuciosa da estrutura da valva aórtica e diâmetros aórticos e investigação de outras anomalias comuns. Mesmo se nenhuma anomalia for detectada e a PA sistêmica for normal, deve-se realizar uma reavaliação do sistema cardiovascular a cada 10 anos, especialmente antes de uma gravidez desejada ou com o desenvolvimento de hipertensão. Se forem encontrados defeitos congênitos, o acompanhamento e o tratamento serão ditados pelo defeito específico. Crianças devem ser transferidas para uma clínica de cardiopatia congênita adulta, pois elas estão em risco contínuo de complicações aórticas durante a idade adulta. Os seguintes algoritmos das diretrizes da American Heart Association para a saúde cardiovascular na síndrome de Turner ajudará na investigação e no acompanhamento.[5][Figure caption and citation for the preceding image starts]: Algoritmo para rastreamento e monitoramento de doença cardiovascular congênita na síndrome de Turner em garotas <15 anosAdaptado de: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].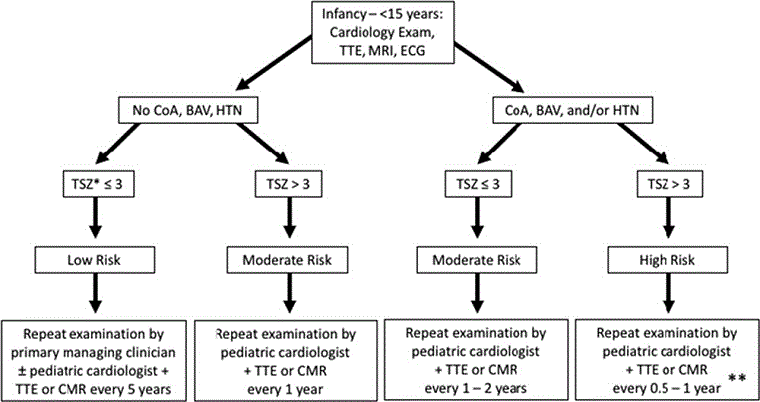 [Figure caption and citation for the preceding image starts]: Algoritmo para rastreamento e monitoramento de doença cardiovascular congênita na síndrome de Turner em mulheres e garotas >15 anos. (CC, cardiopatia congênita; VA, valva aórtica; VAB, valva aórtica bicúspide; DAPVP, drenagem anômala parcial das veias pulmonares; Coarc, coarctação; CCA, cardiopatia congênita adulta; SCV, sistema cardiovascular; HTN, hipertensão)Adaptado de: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].
[Figure caption and citation for the preceding image starts]: Algoritmo para rastreamento e monitoramento de doença cardiovascular congênita na síndrome de Turner em mulheres e garotas >15 anos. (CC, cardiopatia congênita; VA, valva aórtica; VAB, valva aórtica bicúspide; DAPVP, drenagem anômala parcial das veias pulmonares; Coarc, coarctação; CCA, cardiopatia congênita adulta; SCV, sistema cardiovascular; HTN, hipertensão)Adaptado de: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].
O uso deste conteúdo está sujeito ao nosso aviso legal