Etiologia
Não há fatores predisponentes ou causais conhecidos para a síndrome de Turner. A ocorrência é esporádica exceto por casos raros em que uma pequena deleção no cromossomo X pode ser passada de mãe para filha.
A perda de um cromossomo sexual X ou Y inteiro ou de apenas uma parte dele durante a fase inicial da embriogênese geralmente resulta no desenvolvimento de um feto do sexo feminino com anomalias de desenvolvimento. A monossomia do cromossomo Y não é viável. Os fetos 46,XY que perdem o cromossomo Y relativamente tarde durante o desenvolvimento podem ter informação suficiente do cromossomo Y para induzir o desenvolvimento masculino parcial ou completo. No entanto, esses pacientes são identificados como do sexo masculino e são considerados sob o diagnóstico de "distúrbios ou diferenças do desenvolvimento sexual" em vez de síndrome de Turner. Indivíduos do sexo masculino com linhagens celulares 45,X apresentam graus variáveis de baixa estatura, puberdade tardia ou infertilidade e risco de distúrbio cardiovascular.[14]
O fenótipo é determinado pelo momento da perda cromossômica, persistência de linhagens celulares normais (abundância relativa e distribuição tecidual) e patrimônio genético.
Fisiopatologia
Regiões homólogas dos cromossomos sexuais (região pseudoautossômica [PAR]) se comportam como autossomos, com pareamento dos homólogos e recombinação, e contêm genes que escapam da inativação do X. Esses genes são presumivelmente necessários em dose dupla em indivíduos do sexo masculino e feminino. Existem cerca de 20 genes conhecidos e vários outros genes preditos na PAR1, localizada na porção terminal de Xp. Um grupo menor de genes homólogos é encontrado na PAR2, na porção terminal de Xq. A haploinsuficiência para genes PAR1 está envolvida na síndrome de Turner.
[Figure caption and citation for the preceding image starts]: Regiões pseudoautossômicas dos cromossomos X e YDo acervo pessoal de Carolyn Bondy, MS, MD [Citation ends].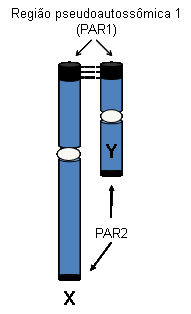
A fisiopatologia específica por trás das anomalias do desenvolvimento observadas na síndrome de Turner é mal compreendida.
O comprometimento do crescimento de ossos longos e do desenvolvimento esquelético ocasionando baixa estatura foi associado à haploinsuficiência para um gene específico do cromossomo sexual, o SHOX, localizado na PAR, o qual codifica para um fator de transcrição expresso em ossos e cartilagens em desenvolvimento.[15][16]
A haploinsuficiência para outros genes pseudoautossômicos causa defeitos cardiovasculares congênitos que induzem à alta mortalidade na síndrome de Turner.[17][18] Dois genes, TIMP1 e TIMP3, que estão ambos localizados no braço curto do cromossomo X quando o hemizigoto aumenta o risco de aortopatia com razão de chances de 12.86.[19]
A causa de insuficiência ovariana prematura permanece incerta, embora genes localizados nos braços curtos e longos do cromossomo X tenham sido implicados.[20] O centro de inativação do X (XIC) está localizado no braço longo, Xq. A deleção do Xq distal ao XIC está mais associada à insuficiência ovariana prematura que a outros achados clínicos da síndrome de Turner. Os ovários se formam normalmente em fetos 45,X do sexo feminino, embora muitos demonstrem morte acelerada de oócitos e degeneração ovariana em fitas fibrosas.[21]
A haploinsuficiência para genes localizados no Xp resulta em caráteres neurocognitivos distintos, como escores superiores para desempenho verbal em comparação a habilidade visuoespacial e dificuldades no reconhecimento de expressões faciais.[22]
Classificação
Classificação citogenética[6]
45,X não mosaico
Cromossomo X único em todas as células somáticas (monossomia do X); compreende cerca de 60% das pacientes com síndrome de Turner.
X ou Y fragmentados
Deleções Xp (46,X,delXp), cromossomos isoXq (46,X,iXq), deleções Xq e cromossomos X ou Y em anel com deleções intersticiais substanciais. O cromossomo isoXq, que consiste na deleção do Xp e fusão de 2 braços longos, é a anomalia estrutural mais comum associada à síndrome de Turner. A deleção de grande parte do Xp também está associada à síndrome. Frequentemente, o cromossomo sexual anormal é perdido em algumas células durante o desenvolvimento embrionário, resultando em um mosaicismo de linhagem celular 45,X em associação com a linhagem 46,X, fragX (ver a figura A abaixo).
45,X mosaico
Há dois tipos de mosaicismo.
A perda de um cromossomo sexual durante as divisões iniciais de células embrionárias pode resultar em uma mistura de células normais 46,XX e células 45,X em proporções variáveis pelos tecidos corporais e inclui aproximadamente 15% das pacientes com síndrome de Turner: por exemplo, 45,X (50%)/46,XX (50%) (ver figura B i. abaixo). A proporção relativa de células normais em tecidos diferentes irá influenciar o fenótipo.
Por outro lado, a formação de um embrião 46,X,abnX frequentemente é acompanhada pela perda do X fragmentado durante algumas divisões celulares embrionárias, resultando em mosaicismo para uma linhagem celular monossômica 45,X juntamente com uma linhagem celular contendo um cromossomo X fragmentado; por exemplo, iXq (ver figura B ii. abaixo). Esse indivíduo não tem células normais, e o fenótipo completo é esperado.[Figure caption and citation for the preceding image starts]: Anormalidades do cromossomo X na síndrome de Turner; ver o texto para explicaçãoDo acervo pessoal de Carolyn Bondy, MS, MD [Citation ends].
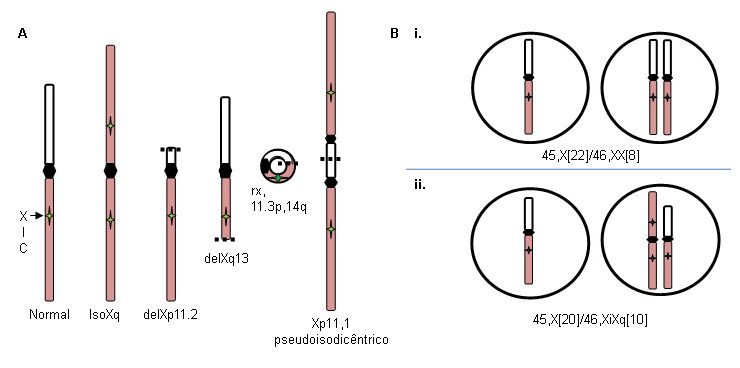
O uso deste conteúdo está sujeito ao nosso aviso legal