Abordagem
Uma anamnese cautelosa, um exame físico meticuloso e um cariótipo em sangue periférico são cruciais para realizar um diagnóstico preciso da síndrome de Turner.
História
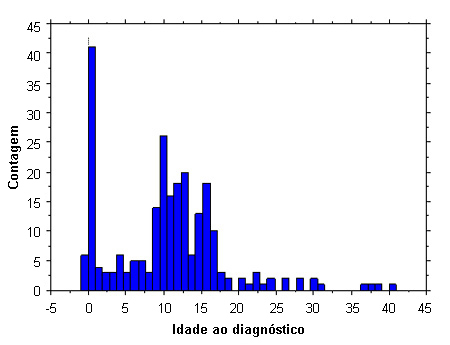
Idade ao diagnóstico
Relativamente poucas pacientes são diagnosticadas durante a primeira infância. O maior número de detecções ocorre entre os 10 e 16 anos de idade, devido à combinação das características de baixa estatura e puberdade tardia. Menos de 10% dos casos são diagnosticados em exame pré-natal e outros 20% são detectados na primeira infância, devido à presença de linfedema, pescoço alado e/ou defeitos cardíacos congênitos. Outros 10% dos casos são diagnosticados na idade adulta, devido à amenorreia secundária. Os perfis de investigação são semelhantes na Europa e nos EUA.[9][10]
[Figure caption and citation for the preceding image starts]: Dados do perfil de investigação da síndrome de Turner; 300 indivíduos do sexo feminino; idade (em anos) ao diagnósticoDo acervo pessoal de Carolyn Bondy, MS, MD (National Institute of Child Health and Human Development natural history study 2001-2007); McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775 [Citation ends].
História de nascimento
Aproximadamente 10% dos neonatos com síndrome de Turner manifestam clinicamente uma cardiopatia congênita grave, como estenose aórtica, coarctação aórtica ou hipoplasia do coração esquerdo. No entanto, muitos outros apresentam defeitos que são clinicamente silenciosos ou sutis, como valva aórtica bicúspide ou conexões venosas pulmonares anômalas parciais.[23] O diagnóstico pode ser óbvio ao nascimento, eventualmente devido a achados clínicos cardíacos ou a linfedema (dorsos dos pés inchados) ou pescoço alado.
Baixa estatura
O baixo crescimento frequentemente é o sintoma primário. Meninas com síndrome de Turner são relativamente baixas desde a primeira infância e, aos 10 anos de idade, geralmente se enquadram abaixo do quinto percentil para altura em curvas de crescimento específicas para idade e sexo. A altura também pode ser avaliada utilizando parâmetros específicos para Turner. Magic Foundation: growth charts Opens in new window
Portanto, a história de crescimento da criança deve ser colhida, focando em causas potenciais de baixa estatura na infância. Uma anamnese detalhada da estatura da família (pais, irmãos) também deve ser obtida. Medições seriadas podem revelar um cruzamento de percentis para baixo, sugerindo uma velocidade de crescimento anormalmente baixa.
O comprimento deve ser medido na posição supina em crianças até os 2 anos de idade e, a partir de então, na posição ortostática. A baixa estatura é proporcional, envolvendo igualmente o tronco e os membros inferiores.
O crescimento da criança está fortemente relacionado ao seu potencial genético, e o deficit de crescimento deve ser avaliado utilizando a predição de altura média parental para identificar a trajetória genética de crescimento da menina. A altura média parental para uma menina é calculada da seguinte forma:
(altura da mãe em centímetros + altura do pai em centímetros)/2.0 a 6.4 cm [(altura da mãe em polegadas + altura do pai em polegadas)/2.0 a 2.5 polegadas].
Puberdade tardia
Aproximadamente 15% das meninas continuam a apresentar tecido ovariano funcional e puberdade espontânea, embora a maioria apresente puberdade tardia e amenorreia primária. Quase todas as meninas sofrem de menopausa prematura.[7]
Outros dados da história médica
Crises frequentes ou extraordinariamente graves de otite média na infância.
Habilidades sociais deficientes.
Exame físico
O exame físico de rotina pode revelar características dismórficas patognomônicas:
[Figure caption and citation for the preceding image starts]: Características patognomônicas da síndrome de TurnerDo acervo pessoal de Carolyn Bondy, MS, MD (com dados de Ullrich O. Z. Kinderheilk. 1930;49:271-76 e Turner HH. Endocrinology. 1938;23:566-74) [Citation ends].
Outras características incluem:
Fissura palpebral oblíqua, ptose ou olhos cobertos
Nevos melanocíticos múltiplos
Unhas hiperconvexas e distróficas
Escoliose.
Estado puberal
Pelos pubianos são tipicamente escassos, e o desenvolvimento puberal é mínimo.
Exame cardiovascular
Sopro ou cliques são sugestivos de doença da valva aórtica.
A pressão arterial (PA) deve ser medida em todos os membros, e os pulsos femorais devem ser palpados. A hipertensão em um ou ambos os membros superiores sugere coarctação aórtica.
A prevalência de cardiopatia congênita é muito maior naquelas com clara evidência de linfedema fetal, que pode ser descrito como edema de tecidos, especialmente da cabeça e pescoço, devido a desenvolvimento linfático deficiente. As manifestações comuns pós-parto são pescoço alado, com orelhas de implantação baixa e da linha do cabelo.
As comorbidades/complicações observadas frequentemente na síndrome de Turner são apresentadas abaixo.
[Figure caption and citation for the preceding image starts]: Defeitos cardiovasculares associados a pescoço aladoDados compilados de Sachdev V, et al. J Am Coll Cardiol. 2008;51:1904-1909, e Loscalzo ML, et al. Pediatrics. 2005;115:732-735 [Citation ends].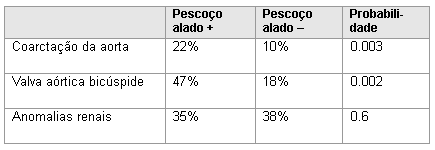 [Figure caption and citation for the preceding image starts]: Comorbidades e complicações em meninas com síndrome de Turner; o diagnóstico de defeitos cardíacos congênitos foi feito por ecocardiografia e angiografia por ressonância magnética cardíaca; as imagens renal e hepática foram feitas por ultrassonografia; a hipertensão foi determinada por monitoramento ambulatorial da pressão arterial (PA) usando padrões baseados em altura; *inclui drenagem anômala parcial das veias pulmonares, arco transversal da aorta alongado, arco aórtico direito; ** >10% de elevação da alanina aminotransferase (ALT) e/ou aspartato transaminase (AST)McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775 [Citation ends].
[Figure caption and citation for the preceding image starts]: Comorbidades e complicações em meninas com síndrome de Turner; o diagnóstico de defeitos cardíacos congênitos foi feito por ecocardiografia e angiografia por ressonância magnética cardíaca; as imagens renal e hepática foram feitas por ultrassonografia; a hipertensão foi determinada por monitoramento ambulatorial da pressão arterial (PA) usando padrões baseados em altura; *inclui drenagem anômala parcial das veias pulmonares, arco transversal da aorta alongado, arco aórtico direito; ** >10% de elevação da alanina aminotransferase (ALT) e/ou aspartato transaminase (AST)McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775 [Citation ends].
Cariótipo
O padrão ouro para diagnóstico nas últimas três décadas tem sido a análise citogenética de cariótipo de sangue periférico e este exame dá suporte à maior parte das observações clínicas e informações prognósticas. Métodos mais novos de análise molecular utilizando sequenciamento de ácido desoxirribonucleico (DNA) ou análises de microarray têm sido aperfeiçoados nos últimos anos, mas ainda não estão amplamente disponíveis.[24][25]
O cariótipo em leucócito periférico com 30 células examinadas é o teste padrão para diagnóstico.[26] Mais de 10% das células apresentam perda de um cromossomo sexual inteiro ou de uma parte significativa dele. Esse exame identifica mosaicismo de 10% ou mais, com 95% de confiança. O exame é indicado em:[27]
Uma mulher com uma ou mais das seguintes características clínicas:
Higroma cístico fetal ou hidropisia, especialmente quando grave
Baixa estatura idiopática
Características faciais específicas
Defeitos cardíacos congênitos obstrutivos do lado esquerdo
Puberdade ou menarca tardia inexplicada
Casal com infertilidade inexplicada
Uma mulher com pelo menos duas das seguintes características clínicas:
Anomalia renal (forma de ferradura, ausência ou hipoplasia)
Deformidade de Madelung
Problemas neuropsicológicos ou psiquiátricos
Nevos melanocíticos ou múltiplos típicos
Unhas displásicas ou hiperconvexas
Outros defeitos cardíacos congênitos
Idade <40 anos com deficiência auditiva e baixa estatura.
Recomenda-se que o teste deva ser repetido nos seguintes casos:[5]
Lactentes diagnosticados antes de nascer devem realizar um cariótipo pós-parto para confirmar o diagnóstico
Indivíduos diagnosticados por swab bucal apenas
Indivíduos com cariótipo realizado no passado distante
Quando o relatório original não está disponível para revisão.
Uma amostra de sangue de 15 mL, coletada usando técnica estéril e mantida à temperatura ambiente, deve ser enviada ao laboratório de genética em até 24 horas. De acordo com o American College of Medical Genetics, no mínimo 20 células devem ser analisadas para contagem de cromossomos durante a busca de possíveis anormalidades cromossômicas sexuais em que mosaicismo é comum. A análise está completa se o mosaicismo for confirmado. Caso seja observada uma célula com perda, ganho ou reorganização do cromossomo sexual nas 20 primeiras células analisadas, pelo menos 10 células adicionais devem ser avaliadas.[28][29]
Em pacientes com forte suspeita do diagnóstico, se o exame comprovar <10% de células anormais, mais metáfases deverão ser contadas juntamente com hibridização in situ por fluorescência (FISH) ou outros tipos celulares deverão ser analisados com FISH de interfase, em consulta a um geneticista e/ou citogeneticista.[30] Com base na citogenética, as pacientes com síndrome de Turner podem ser classificadas como:
45,X não mosaico
Cromossomo X único em todas as células somáticas (monossomia do X); compreende cerca de 60% das pacientes com síndrome de Turner.
X ou Y fragmentados
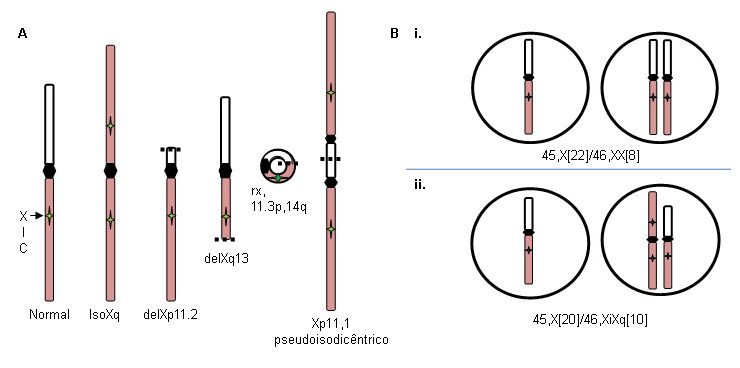
Deleções Xp (46,X,delXp), cromossomos isoXq (46,X,iXq), deleções Xq e cromossomos X ou Y em anel com deleções intersticiais substanciais. O cromossomo isoXq, que consiste na deleção do Xp e fusão de dois braços longos, é a anomalia estrutural mais comum associada à síndrome de Turner. A deleção de grande parte do Xp também está associada à síndrome. Frequentemente, o cromossomo sexual anormal é perdido em algumas células durante o desenvolvimento embrionário, resultando em um mosaicismo de linhagem celular 45,X em associação com a linhagem 46,X, fragX (ver a figura A abaixo).
A detecção de um cromossomo marcador ou em anel requer análises futuras com FISH com marcadores específicos para Y. Pacientes com um cromossomo Y evidente requerem avaliação adicional em vista do risco de gonadoblastoma.
45,X mosaico
Existem dois tipos de mosaicismo.
A perda de um cromossomo sexual durante as divisões iniciais de células embrionárias pode resultar em uma mistura de células normais 46,XX e células 45,X em proporções variáveis pelos tecidos corporais e inclui cerca de 15% das pacientes com síndrome de Turner; por exemplo, 45,X (50%)/46,XX (50%) (ver figura B i. abaixo). A proporção relativa de células normais em tecidos diferentes irá influenciar o fenótipo.
Por outro lado, a formação de um embrião 46,X,abnX frequentemente é acompanhada pela perda do X fragmentado durante algumas divisões celulares embrionárias, resultando em mosaicismo para uma linhagem celular monossômica 45,X juntamente com uma linhagem celular contendo um cromossomo X fragmentado; por exemplo, iXq (ver figura B ii. abaixo). Esse indivíduo não tem células normais, e o fenótipo completo é esperado.
[Figure caption and citation for the preceding image starts]: Anormalidades do cromossomo X na síndrome de Turner; ver o texto para explicaçãoDo acervo pessoal de Carolyn Bondy, MS, MD [Citation ends].
Investigações subsequentes
Idade óssea
A radiografia é o método usado para avaliar maturação esquelética. Tipicamente, há um retardo leve (2 anos a menos que a idade cronológica). Recomenda-se a avaliação do potencial de crescimento em todas as meninas pré-púberes com a síndrome.
Hormônio folículo-estimulante (FSH) e hormônio antimülleriano (HAM) séricos[5]
FSH elevado e níveis muito baixos ou indetectáveis de HAM são preditores de falência ovariana completa. No entanto, o potencial ovariano não pode ser predito com certeza em meninas jovens, já que em meninas com falência ovariana, os níveis de FSH se elevam à faixa da menopausa apenas na idade da puberdade normal.
Ultrassonografia pélvica
Identifica um útero imaturo e morfologia ovariana em fita.
Realizada em meninas mais velhas e em mulheres.
Os ovários se formam normalmente em fetos 45,X do sexo feminino, embora muitos demonstrem morte acelerada de oócitos e degeneração ovariana em fitas fibrosas.[21]
Radiografia do esqueleto
Realizada na infância, aos 5-6 anos e 12-14 anos, para avaliar anomalias associadas, como deformidades do punho e escoliose.[5]
A deformidade de Madelung (ulna distal proeminente) é encontrada em apenas aproximadamente 5% das pacientes, mas é comum a ocorrência de graus menores de anomalias do punho que podem causar problemas funcionais.
Exames de sangue basais para rastrear comorbidades/complicações[5]
Testes de função tireoidiana (TFTs) para doença tireoidiana autoimune em todos os pacientes no momento do diagnóstico. Anticorpos antitireoidianos podem ser medidos se os TFTs forem anormais.
TFHs para descartar "hepatite de Turner" se tiver 10 anos de idade ou mais.
Glicemia de jejum e hemoglobina glicada (HbA1c) para identificar risco de diabetes se tiver 10 anos de idade ou mais.
Lipídios séricos para evidência de dislipidemia, se tiver 18 anos de idade ou mais, e se houver pelo menos um fator de risco para doença cardiovascular (verifique também as recomendações regionais).
Nível de imunoglobulina A (IgA) e IgA antitransglutaminase tecidual para rastreamento de doença celíaca, se tiver 2 anos.
Níveis de vitamina D verificados após os 9 anos de idade.
Além disso, no momento do diagnóstico, os seguintes testes devem ser realizados:
Rastreamento para perda auditiva condutiva e/ou neurossensorial[5]
Uma avaliação audiológica formal deve ser realizada para assegurar medições técnicas precoces e adequadas e outras medidas de reabilitação.
Rastreamento de erros refrativos e anormalidades visuais[5]
Estrabismo, ambliopia, hiperopia e miopia, ptose e cegueira de cores foram relatados. Mulheres com síndrome de Turner têm uma dobra epicântica (dobra na pálpebra superior que cobre o canto interno do olho) e hipertelorismo (maior distância entre os olhos). Um exame oftalmológico abrangente deve ser realizado entre 12 e 18 meses de idade, ou no momento do diagnóstico, se ocorrer em idade mais avançada.
Rastreamento para anomalias renais congênitas
Uma ultrassonografia renal deve ser realizada para avaliar a presença de anomalias estruturais, como rim em ferradura, agenesia renal e sistema coletor duplicado, que afetam aproximadamente 25% das pacientes com síndrome de Turner.[3][4][5] A função renal geralmente é normal, mas a obstrução do sistema coletor está associada à infecção urinária e pode requerer correção.
Rastreamento para defeitos cardiovasculares congênitos
No momento do diagnóstico , independente da idade, uma avaliação cardiovascular completa dever ser realizada por um especialista em cardiopatia congênita.[27]
Deve-se tentar visualizar a valva aórtica, a aorta torácica e as veias pulmonares por ecocardiografia transtorácica (ETT) em lactentes ou por ressonância nuclear magnética cardíaca (RNMC) ou TC em crianças mais velhas e adultos. A RNMC requer sedação em pacientes jovens e, portanto, se as avaliações clínica e ecocardiográfica parecerem normais, é razoável esperar até que a criança tenha idade suficiente para cooperar sem sedação (geralmente 9-10 anos) para realizar um rastreamento por RNMC.
Um ECG deve ser realizado para avaliar potenciais anormalidades de condução e repolarização.
Se forem encontrados defeitos congênitos, o acompanhamento e o tratamento serão ditados pelo defeito específico.[27] Crianças com defeitos cardiovasculares devem ser transferidas para uma cirurgia de cardiopatia congênita adulta, pois elas estão em risco contínuo de complicações aórticas durante a idade adulta.
[Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética (RNM) cardíaca revelando arco aórtico normal em formato de bengala à esquerda, comparado com uma coarctação aórtica não diagnosticada anteriormente, imediatamente após a origem da artéria subclávia esquerda (seta), detectada por RNM em uma mulher adulta com síndrome de Turner com hipertensão grave dos membros superioresDo acervo pessoal de Carolyn Bondy, MS, MD (estudo do National Institutes of Health [NIH]) [Citation ends].
A coarctação aórtica e as valvas aórticas bicúspides são as anormalidades mais comuns. Outras anormalidades incluem drenagem anômala parcial das veias pulmonares, hipoplasia do coração esquerdo e dilatação aórtica. A prevalência de defeitos cardiovasculares é muito maior naquelas com clara evidência de linfedema fetal, como pescoço alado. Uma valva bicúspide funcional como resultado da fusão completa ou parcial dos folhetos coronarianos direito e esquerdo é observada em 30% das pacientes assintomáticas e representa um risco de infecção, deterioração valvar e dilatação e dissecção aórtica.[18] Defeitos cardiovasculares congênitos constituem a principal causa de mortalidade prematura na síndrome de Turner. Dois genes, TIMP1 e TIMP3, que estão ambos localizados no braço curto do cromossomo X quando o hemizigoto aumenta o risco de aortopatia com razão de chances de 12.86.[19] Uma coarctação aórtica será revelada na angiografia por RM (ARM) como uma estenose focal em oposição a uma aparência aórtica normal de um arco aórtico em formato de bengala.
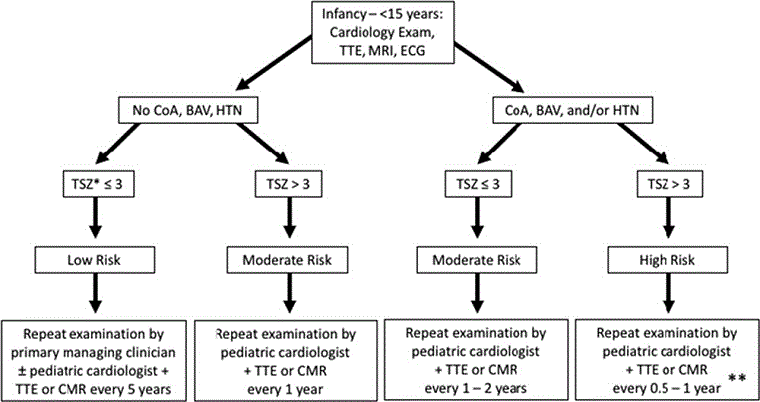
Algoritmos baseados nas diretrizes da American Heart Association para a saúde cardiovascular na síndrome de Turner ajudará na investigação e no acompanhamento.[27]
[Figure caption and citation for the preceding image starts]: Algoritmo para rastreamento e monitoramento de doença cardiovascular congênita na síndrome de Turner em garotas <15 anosAdaptado de: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].
[Figure caption and citation for the preceding image starts]: Algoritmo para rastreamento e monitoramento de doença cardiovascular congênita na síndrome de Turner em mulheres e garotas >15 anos. (CC, cardiopatia congênita; VA, valva aórtica; VAB, valva aórtica bicúspide; DAPVP, drenagem anômala parcial das veias pulmonares; Coarc, coarctação; CCA, cardiopatia congênita adulta; SCV, sistema cardiovascular; HTN, hipertensão)Adaptado de: Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].

O uso deste conteúdo está sujeito ao nosso aviso legal