ეტიოლოგია
მწვავე ლიმფობლასტური ლეიკემიის (ALL) მიზეზი უცნობია. გარკვეული ფაქტორები ასოცირდება ALL-ის განვითარებასთან.
გენეტიკური
ALL-ის დიაგნოზი მონოზიგოტურ ტყუპში (<6 წლის ასაკში) დაკავშირებულია 10%-დან 15%-მდე ალბათობასთან, რომ მეორე ტყუპს განუვითარდეს ALL.[11]
ALL ასოცირდება ტრიზომია 21-თან, კლაინფელტერის სინდრომთან და მემკვიდრეობით დაავადებებთან ჭარბი ქრომოსომული მყიფეობით, როგორიცაა ფანკონის ანემია, ბლუმის სინდრომი და ატაქსია-ტელანგიექტაზია.[1][2][11][12][13]
არსებობს მზარდი მტკიცებულება, რომელიც მიუთითებს ჩანასახის მიდრეკილებაზე ALL-ის მიმართ.[14][15][16][17][18] ALL-თან დაკავშირებული ჩანასახის მუტაციები დაფიქსირებულია ALL-ით დაავადებული ბავშვების დაახლოებით 4%-ში.[14]
ეკოლოგიური: რადიაციის ზემოქმედებისა და მოწევის ჩათვლით.[1][3][19]
მკურნალობა ქიმიოთერაპიით.[19]
ფოლიუმის მჟავის მეტაბოლიზმის პოლიმორფიზმი ასოცირებული იყო ALL-ის რისკთან შემთხვევის კონტროლის კვლევებში.[24][25][26][27]
პათოფიზიოლოგია
ALL-ში, ლიმფოიდური პროგენიტორული უჯრედის გენეტიკური დარღვევები იწვევს უკონტროლო პროლიფერაციას და კლონურ გაფართოებას. ლეიკემიური ლიმფობლასტები შედიან ძვლის ტვინში და სხვა ორგანოებში, რაც არღვევს მათ ნორმალურ ფუნქციას. ლეიკემიური ლიმფობლასტები ასევე შეიძლება ცირკულირებდნენ სისხლში.
ლეიკემიური ლიმფობლასტები წარმოადგენს ერთი უჯრედის კლონურ გაფართოებას.[2][28][29][30]
ლეიკემიური ლიმფობლასტები იმეორებენ ლიმფოიდური წინამორბედი უჯრედის მახასიათებლებს. ALL-ის გენეტიკური დარღვევები მოიცავს ქრომოსომულ გადანაწილებას (მაგ., ტრანსლოკაციას), ანევპლოიდიას (ქრომოსომების არანორმალური რაოდენობა) და სხვა გენეტიკურ მუტაციებს. ქრომოსომული ტრანსლოკაცია ან ანევპლოიდი გვხვდება შემთხვევების 75%-ში. გადაადგილებები ჩვეულებრივ განმეორებადია და იშვიათად კლასიფიცირდება როგორც შემთხვევითი გადაადგილებები.[29][30][31][32]
ფილადელფიის ქრომოსომაზე დადებითი B-ALL (Ph+ B-ALL)
ერთ-ერთი ყველაზე გავრცელებული და კლინიკურად მნიშვნელოვანი ქრომოსომული ტრანსლოკაცია მოზრდილებში B-ALL არის t(9;22)(q34;q11), რაც იწვევს BCR::ABL1 შერწყმის გენს 22 ქრომოსომაზე (ანუ ფილადელფიის ქრომოსომა).[33] BCR:: ABL1 შერწყმის გენი აკოდირებს აქტიურ ტიროზინ კინაზას, რომელიც გარდაქმნის ნორმალურ ჰემატოპოეზურ ღეროვან უჯრედებს ავთვისებიან უჯრედებად.[Figure caption and citation for the preceding image starts]: BCR::ABL1 ტრანსლოკაციადოქტორ ჰან მიინტისა და დოქტორ რობერტ ჩენის კოლექციიდან; გამოიყენება ნებართვით [Citation ends].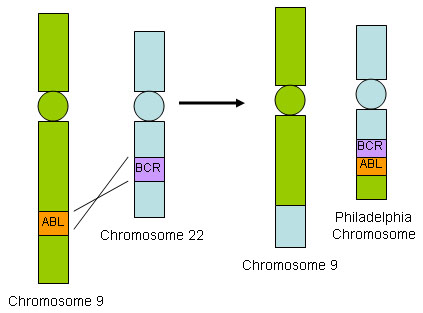
ფილადელფიის მსგავსი B-ALL (Ph-ის მსგავსი B-ALL)
Ph-ის მსგავსი B-ALL არის მაღალი რისკის ქვეტიპი, რომელსაც აქვს Ph+ B-ALL-ის მსგავსი გენის გამოხატვის პროფილი, მაგრამ არ გააჩნია BCR::ABL1 შერწყმის გენი (ფილადელფიის ქრომოსომა).[33] Ph-ის მსგავსი B-ALL შეადგენს სტანდარტული და მაღალი რისკის მქონე ბავშვთა B-ALL შემთხვევების 10% და 13%-ს, შესაბამისად.[29]
Ph-ის მსგავსი B-ALL-ის სიხშირე იზრდება ასაკთან ერთად, რაც შეადგენს ახალგაზრდების შემთხვევების >25%-ს.[33] Ph-ის მსგავსი B-ALL-ის არსებობა დაკავშირებულია არახელსაყრელ შედეგთან.[34][35]
Ph-ის მსგავსი B-ALL პაციენტები შეიძლება დაიყოს შემდეგნაირად:[36]
ტიპი I, ABL კლასის შერწყმა (ABL1, ABL2, CSF1R, PDGFRB)
ტიპი II, ერითროპოეტინის რეცეპტორი (EPOR) ან JAK2 გადაწყობები
ტიპი III, ციტოკინის რეცეპტორის მსგავსი ფაქტორი 2 (CRLF2) გადაწყობები (ხშირად თან ახლავს JAK2 მუტაციებს და JAK-STAT სიგნალის აქტივაციას)
ტიპი IV, სხვა მუტაციები, რომლებიც ააქტიურებს JAK-STAT სიგნალის გზას (IL7R, FLT3, SH2B3, TYK2, IL2RB)
ტიპი V, კინაზას იშვიათი სხვა მუტაციები (NTRK3, DGKH)
ტიპი VI, RAS-გზის მუტაციები (KRAS, NRAS, PTPN11, NF1)
ტიპი VII, კინაზას გენებში მუტაციები არ ვლინდება.
სხვა გენეტიკური დარღვევები
FLT3 და NOTCH1 იდენტიფიცირებულია, როგორც შერეული ხაზის ლეიკემიის (MLL)/ჰიპერდიპლოიდური და T-ALL მუტაციის მქონე გენები, შესაბამისად.[37] CREBBP მუტაციები ვლინდება რეციდივის შემთხვევათა 18%-ში და შეიძლება ნიშნავდეს რეზისტენტობას მკურნალობის მიმართ.[38] PAX5 გენი მუტირებულია მწვავე ლიმფოციტური ლეიკემიის მქონე პედიატრიული პაციენტების 30%-ში.[39][40] PHF6 მუტაციები ვლინდება ზრდასრულთა T-მწვავე ლიმფოციტური ლეიკემიის შემთხვევების 38%-ში.[41] CDKN2A მუტაციები ვლინდება T-მწვავე ლიმფოციტური ლეიკემიის შემთხვევების 42%-ში.[42]
ზოგიერთი გენის გადაწყობამ შეიძლება გამოიწვიოს მუტაციების ფუნქციის დაქვეითება ან მომატება, რომელიც ეხება ტრანსკრიფციის ფაქტორებს, და თამაშობენ დიდ როლს ჰემატოპოეზის განვითარებაში. ასეთი გენის გადაწყობის მაგალითია t(12;21)(p13;q22) ქრომოსომული ტრანსლოკაცია, რომელიც იწვევს შერწყმის გენს ETV6::RUNX1 (ასევე ცნობილია როგორც TEL::AML1).[1][3]
სიმსივნე-სუპრესორული გენების დაკარგვა ან ინაქტივაცია წაშლისა და გენის გადაწყობის გზით (მაგ., IKZF1, p16INK4) ასოცირდება ALL-ის განვითარებასთან.[43][44] IKZF1 მუტაციები შეიძლება იყოს რეციდივის პროგნოზის მაჩვენებელი[45] IKZF1-ის წაშლა CDKN2A-ში, CDKN2B, PAX5 ან PAR1-ში (ERG წაშლის არარსებობის შემთხვევაში) თანმხლები წაშლით განსაზღვრავს ქვეჯგუფს, რომელსაც მოიხსენიებენ როგორც 'IKZF1 plus', რომელიც დაკავშირებულია განსაკუთრებით ცუდ პროგნოზთან.[46]
ზოგიერთი მორეციდივე გენეტიკური პათოლოგია (მაგ. BCR::ABL1, KMT2A გადაწყობა, ETV6::RUNX1) ჩართულია დაავადების კლასიფიკაციის სისტემებში ჯანმრთელობის მსოფლიო ორგანიზაციის (WHO) და საერთაშორისო კონსენსუსის კლასიფიკაციის (ICC) ALL-ის ქვეკლასიფიკაციისთვის.[6][7] იხილეთ კლასიფიკაცია.
კლასიფიკაცია
ჯანდაცვის მსოფლიო ორგანიზაციის მე-5 გამოცემა ჰემატოლიმფოიდური სიმსივნეების კლასიფიკაციის: ლიმფოიდური ნეოპლაზმები[6]
ALL-ის (და ერთეულების) კლასიფიკაცია ეფუძნება გვარს (B-ALL ან T-ALL) და ციტოგენეტიკური/მოლეკულური ანომალიების არსებობას.
B-უჯრედოვანი ლიმფობლასტური ლეიკემიები/ლიმფომები:
B-ლიმფობლასტური ლეიკემია/ლიმფომა, სხვაგვარად არ არის მითითებული
B-ლიმფობლასტური ლეიკემია/ლიმფომა მაღალი ჰიპერდიპლოიდიით
B-ლიმფობლასტური ლეიკემია/ლიმფომა ჰიპოდიპლოიდიით
B-ლიმფობლასტური ლეიკემია/ლიმფომა iAMP21-ით
B-ლიმფობლასტური ლეიკემია/ლიმფომა BCR::ABL1 შერწყმით
B-ლიმფობლასტური ლეიკემია/ლიმფომა BCR::ABL1-ის მსგავსი მახასიათებლებით
B-ლიმფობლასტური ლეიკემია/ლიმფომა KMT2A გადაწყობით
B-ლიმფობლასტური ლეიკემია/ლიმფომა ETV6::RUNX1 შერწყმით
B-ლიმფობლასტური ლეიკემია/ლიმფომა ETV6::RUNX1-ის მსგავსი მახასიათებლებით
B-ლიმფობლასტური ლეიკემია/ლიმფომა TCF3::PBX1 შერწყმით
B-ლიმფობლასტური ლეიკემია/ლიმფომა IGH::IL3 შერწყმით
B-ლიმფობლასტური ლეიკემია/ლიმფომა TCF3::HLF შერწყმით
B-ლიმფობლასტური ლეიკემია/ლიმფომა სხვა განსაზღვრული გენეტიკური ანომალიებით (მაგ., DUX4, MEF2D, ან ZNF384 გადაკეთებები)
T-ლიმფობლასტური ლეიკემია/ლიმფომა:
T-ლიმფობლასტური ლეიკემია/ლიმფომა, სხვაგვარად არ არის მითითებული
ადრეული T- წინამორბედი ლიმფობლასტური ლეიკემია/ლიმფომა
მიელოიდური ნეოპლაზმებისა და მწვავე ლეიკემიების საერთაშორისო კონსენსუსის კლასიფიკაცია (ICC).[7]
ALL-ის (და ერთეულების) კლასიფიკაცია ეფუძნება გვარს (B-ALL ან T-ALL) და ციტოგენეტიკური/მოლეკულური ანომალიების არსებობას.
B-უჯრედოვანი მწვავე ლიმფობლასტური ლეიკემია/ლიმფომა
B-ALL მორეციდივე გენეტიკური ანომალიებით
B-ALL t(9;22)(q34.1;q11.2)/BCR::ABL1
მხოლოდ ლიმფოიდური ჩართულობით
მრავალხაზოვანი ჩართულობით
B-ALL t(v;11q23.3)/KMT2A-ით გადააწყო
B-ALL t(12;21)(p13.2;q22.1)/ETV6::RUNX1-ით
B-ALL, ჰიპერდიპლოიდური
B-ALL, დაბალი ჰიპოდიპლოიდური
B-ALL, ჰაპლოიდთან ახლოს
B-ALL t(5;14)(q31.1;q32.3)/IL3::IGH
B-ALL t(1;19)(q23.3;p13.3)/TCF3::PBX1-ით
B-ALL, BCR::ABL1-ის მსგავსი, ABL-1 კლასის გადაწყობა
B-ALL, BCR::ABL1-ის მსგავსი, JAK-STAT გააქტიურებულია
B-ALL, BCR::ABL1-ის მსგავსი, სხვაგვარად არ არის მითითებული
B-ALL iAMP21-ით
B-ALL MYC გადაწყობით
B-ALL DUX4 გადაწყობით
B-ALL MEF2D გადაწყობით
B-ALL ZNF384(362) გადაწყობით
B-ALL NUTM1 გადაწყობით
B-ALL HLF გადაწყობით
B-ALL UBTF-ით::ATXN7L3/PAN3,CDX2 ('CDX2/UBTF')
B-ALL მუტაციური IKZF1 N159Y-ით
B-ALL მუტაციური PAX5 P80R-ით
B-ALL, სხვაგვარად არ არის მითითებული
დროებითი ერთეული: B-ALL, ETV6::RUNX1-ის მსგავსი
დროებითი ერთეული: B-ALL, PAX5 ცვლილებით
დროებითი ერთეული: B-ALL, მუტაციით ZEB2 (p.H1038R)/IGH::CEBPE
დროებითი ერთეული: B-ALL, ZNF384 გადაწყობილი მსგავსი
დროებითი ერთეული: B-ALL, KMT2A გადაწყობილი მსგავსი
T-უჯრედოვანი მწვავე ლიმფობლასტური ლეიკემია/ლიმფომა
ადრეული T-უჯრედების წინამორბედი ALL BCL11B გადაწყობით
ადრეული T-უჯრედების წინამორბედი ALL, სხვაგვარად არ არის მითითებული
T-ALL, სხვაგვარად არ არის მითითებული
დროებითი ერთეული: T-ALL, HOXA დაქვეითებული
დროებითი ერთეული: T-ALL, SPI1 rearrangement
დროებითი ერთეული: T-ALL, TLX1 rearrangement
დროებითი პირი: T-ALL, TLX3 rearrangement
დროებითი ერთეული: T-ALL, NKX2 rearrangement
დროებითი ერთეული: T-ALL, TAL1-2 rearrangement
დროებითი ერთეული: T-ALL, LMO1-2 rearrangement
დროებითი ერთეული: T-ALL, BHLH, სხვა
ამ მასალის გამოყენება ექვემდებარება ჩვენს განცხადებას