Etiología
La deficiencia de AVP (AVP-D; anteriormente conocida como diabetes insípida central) surge de una deficiencia absoluta o relativa de arginina vasopresina (AVP); la resistencia a la AVP (AVP-R; anteriormente conocida como diabetes insípida nefrogénica) es el resultado de una insensibilidad renal o resistencia a la AVP. Ambos mecanismos reducen la permeabilidad de los conductos dentro de la nefrona al agua, reduciendo la reabsorción de agua y aumentando así la pérdida de agua renal. Las causas de la AVP-D o la AVP-R pueden ser genéticas o adquiridas.[7]
AVP-D - adquirida
Cirugía de la hipófisis: la cirugía transesfenoidal de la hipófisis, generalmente para un adenoma hipofisario, es una causa frecuente de AVP-D.[3] Las lesiones hipofisiarias se presentan con escasa frecuencia con AVP-D. Las excepciones son el craneofaringioma, las lesiones del tallo hipotalámico/hipofisario infiltrantes o las metástasis de la hipófisis.[3] La AVP-D puede ser transitoria o permanente. Después de la cirugía de la hipófisis, en raras ocasiones puede manifestarse como una respuesta clásica de triple fase: AVP-D inicial aguda, seguida de una fase antidiurética transitoria (que se presenta como hiponatremia debido al síndrome de antidiuresis inadecuada [SIAD]), que avanza posteriormente a AVP-D permanente.[12]
Craneofaringioma: a diferencia de la mayoría de los tumores intracraneales, la AVP-D es relativamente frecuente en pacientes con craneofaringioma, tanto preoperatoriamente (8-35%), como postoperatoriamente (70-90%).[13] Los pacientes con craneofaringiomas también son más propensos a presentar alteraciones asociadas a la sed primaria y postoperatoria, lo que puede exacerbar el equilibrio de fluidos y los problemas de electrolitos.[14]
Lesión craneal postraumática: la AVP-D es frecuente después de un traumatismo craneoencefálico.[15] Es más frecuente que esta sea transitoria, desarrollándose en un 21 a 26% de los casos.[16][17] Se ha notificado AVP-D permanente en aproximadamente el 7% de los casos.[16]
Lesiones del tallo hipofisario: la afectación del tallo pituitario es común en la histiocitosis de células de Langerhans y se ha notificado una AVP-D en hasta el 24% de los pacientes.[18] Otras afecciones infiltrantes del tallo pituitario que pueden producir una AVP-D son el germinoma, las metástasis intracraneales, las enfermedades granulomatosas (sarcoidosis y tuberculosis), infundibulohipofisitis linfocítica y enfermedad relacionada con IgG4.[19][20][21][22][23][24][25][26] Los niños y los adultos jóvenes con AVP-D idiopática y que presentan engrosamiento del tallo hipofisario en el momento de su presentación suelen manifestar una reconocida patología/diagnóstico más amplio con un seguimiento a largo plazo.[27][28]
Enfermedades autoinmunitarias: la AVP-D se asocia a enfermedades autoinmunitarias poliendocrinas, incluida la tiroiditis de Hashimoto y la diabetes mellitus de tipo 1.[29] La AVP-D se ha asociado, en un porcentaje de los casos, con autoanticuerpos a células secretoras de AVP (AVPcAb).[29]
Infecciones del sistema nervioso central: la AVP-D puede desarrollarse como una complicación tardía de la meningitis o la encefalitis.[4][22][23][30][31]
La hemorragia subaracnoidea que afecta a la arteria comunicante anterior, la cual riega el hipotálamo anterior puede dar lugar a una AVP-D y a anomalías en la sed.[32]
Medicamentos: la fenitoína se informa como una causa de AVP-D, al igual que la temozolamida.[33][34]
Una causa infrecuente de AVP-D es el envenenamiento por monóxido de carbono.[35][36]
AVP-D - congénito/hereditario
La AVP-D familiar representa aproximadamente del 1% al 5% de todos los casos de AVP-D. Suele ser una enfermedad hereditaria autosómica dominante debida a una mutación en el gen AVP-NPII localizado en el cromosoma 20p135. Esta afección a menudo hace que muchos miembros de la familia presenten AVP-D clínica en una etapa temprana de la vida.[37]
Las malformaciones congénitas que implican a la hipófisis o al hipotálamo y los defectos de la línea media del cerebro prosencéfalo pueden dar lugar a AVP-D.[38]
La AVP-D puede producirse como un componente del síndrome de Wolfram (SW), un trastorno neurodegenerativo autosómico recesivo y progresivo caracterizado por diabetes mellitus y atrofia óptica, con frecuencia variable de AVP-D y de sordera neurosensorial.[39] Los estudios genéticos han identificado dos subtipos del síndrome de Wolfram (SW).[40] El SW1 lo causan mutaciones de pérdida de función en el gen WFSI del cromosoma 4p16. El WFSI codifica una glicoproteína de aminoácido 890, wolframina, que regula el estrés del retículo endoplásico y la homeostasis del calcio. La pérdida de la función del wolframina desencadena la apoptosis y la muerte celular. Las mutaciones no inactivas del WFSI están asociadas con la pérdida auditiva neurosensorial autosómica dominante. Por lo tanto, el WSI puede representar un extremo de un trastorno del espectro. El SW2 se caracteriza también por la atrofia óptica y la diabetes mellitus. El SW2 lo causan mutaciones de pérdida de función en el gen CISD2 que codifica una proteína expresada, tanto en el retículo endoplasmático, como en la membrana mitocondrial externa. La pérdida de función del CISD2 interrumpe el flujo de calcio, provocando la autofagia y la muerte celular.[39]
AVP-R - adquirida
Medicamentos: la terapia con litio es una causa frecuente de AVP-R. Se ha informado en hasta un 40% de los pacientes que reciben tratamiento con litio a largo plazo, pero se ha informado que la incidencia de este efecto adverso es tan alta como un 85%.[9][10] Se debe al efecto del litio en la movilización de los canales de acuaporina. La afección puede ser irreversible y permanecer después de suspender el tratamiento con litio.[41] Otros fármacos que pueden precipitar la AVP-R pueden ser la demeclociclina, el cisplatino, la colchicina, la gentamicina, la rifampicina y el sevoflurano.[5][42]
Enfermedad sistémica, desequilibrio electrolítico y uropatía postobstructiva: la enfermedad renal crónica, la sarcoidosis renal y la amiloidosis renal son causas reconocidas de AVP-R.[5][21] La AVP-R puede estar causada por hipercalcemia e hipopotasemia, y esto puede revertirse mediante la normalización de los electrolitos.[5] La obstrucción ureteral también es un factor de riesgo reconocido para la AVP-R.[5] Se cree que el aumento de la presión hidrostática suprime la expresión de AQP2 y esta regulación a la baja persiste hasta 30 días después de la liberación de la obstrucción, lo que puede explicar la diuresis que se observa a menudo después de la liberación de la obstrucción grave en los pacientes.[10]
AVP-D - hereditaria
Las mutaciones más comunes (90%) son las del receptor AVPR2, que median la acción antidiurética de la AVP en el conducto colector. Las mutaciones de pérdida de función en el receptor AVPR2 resultan en la AVP-R familiar recesiva ligada al cromosoma X.[5][10][43]
La acuaporina-2 (AQP2) es el canal de agua dependiente de la AVP que hace que el conducto colector distal sea permeable al agua y permite la reabsorción de agua renal distal mediada por la AVP. Las mutaciones de pérdida de función en el gen AQP2 producen AVP-R familiar autosómica recesiva.[5][10][43]Mutaciones de pérdida de función en el gen transportador de urea-B también producen la AVP-R autosómica recesiva.[37]
Embarazo y AVP-D
Los umbrales osmóticos para la sed y la liberación de AVP se ven alterados durante el embarazo.[44] También aumenta 4 veces la depuración metabólica de AVP debido a la producción placentaria de la vasopresinasa/oxitocinasa. Esto puede desenmascarar una AVP-D no reconocida previamente. Además, el embarazo puede agravar la gravedad de cualquier AVP-R o AVP-D existente.[1][6][45] Se han notificado casos de AVP-D gestacional transitoria en pacientes con preeclampsia, síndrome de HELLP (hemólisis, enzimas hepáticas elevadas y recuento bajo de plaquetas) y esteatosis hepática aguda debido a la alteración de la degradación hepática de la vasopresinasa.
Fisiopatología
El volumen de plasma y la osmolalidad se mantienen dentro de un rango estrecho mediante un sistema neurohumoral integrado que coordina el equilibrio entre la bebida dirigida por la sed (ingesta de líquidos) y la pérdida de agua renal (salida de líquidos).[46][47]
El principal regulador de la pérdida de agua renal es la hormona péptida de 9 aminoácidos arginina vasopresina (AVP), también conocida como hormona antidiurética (HAD).[46] La AVP se sintetiza en las neuronas magnocelulares dentro de los núcleos supraópticos (SON) y paraventriculares (PVN) del hipotálamo. La AVP se produce a partir de un péptido precursor de gran tamaño. La transcripción primaria sufre un importante procesamiento postraduccional cuando se produce el tráfico a través de los axones de la neurona magnocelular que terminan dentro de la hipófisis posterior, donde se almacena y libera la hormona. Cada molécula de AVP se cosecreta con dos fragmentos del precursor original: la copeptina y la neurofisina II (NPII).[48] El procesamiento se describe en el siguiente diagrama.
[Figure caption and citation for the preceding image starts]: Vasopresina: estructura genética y procesamiento postraduccionalDe la colección personal del Dr. Stephen Ball [Citation ends].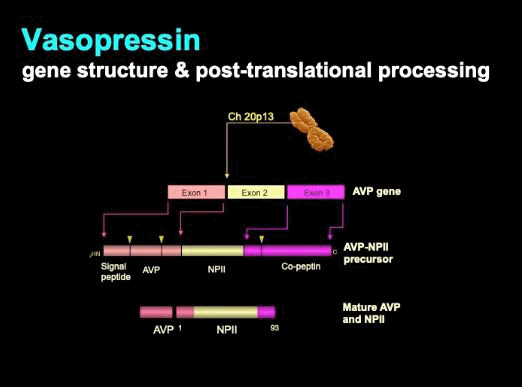
Existe una relación lineal entre la concentración de plasma AVP y la osmolalidad del plasma. La osmolalidad plasmática la detectan los osmoreceptores dentro del hipotálamo y estos aferentes sensoriales regulan la síntesis y liberación de AVP. La liberación de AVP también la regula el volumen circulante y la presión sanguínea. Esta barorregulación puede conducir a la producción y liberación de AVP en las osmolalidades plasmáticas por debajo del umbral osmolar estándar de alrededor de 290 mmol/kg (290 mOsm/kg).[46][47]
[Figure caption and citation for the preceding image starts]: Fisiología de la AVP y la sedDe la colección personal del Dr. Stephen Ball [Citation ends].
La estimulación hiperosmolar graduada define muy claramente las características fisiológicas de la producción de AVP osmorregulada y la sed, representadas en estas dos figuras. La relación lineal entre la osmolalidad del plasma y la AVP del plasma se caracteriza además por un umbral osmolar funcional para la liberación de AVP; y una pendiente característica, que sirve para definir la sensibilidad de la respuesta AVP a la estimulación osmolar. Un conjunto de características muy similares describen la relación entre la sed y la osmolalidad plasmática. Aunque las características de la liberación de AVP osmorregulada y la sed varían entre los individuos (el rango representado aquí como las áreas sombreadas o el nomograma), las características permanecen notablemente constantes dentro de un individuo (representado aquí como la línea sólida dentro del normograma) a lo largo del tiempo.
La AVP actúa sobre los receptores AVP de tipo 2 (AVPR2) en la superficie intersticial de las células del conducto colector distal renal, aumentando la permeabilidad del agua mediante la mayor síntesis de los canales de agua de la acuaporina-2.[46] Éstos se ensamblan e insertan en la membrana apical de las células del conducto colector, lo que da lugar a la reabsorción de agua dependiente de la AVP y a la concentración de orina.
La AVP-D es el resultado de cualquier afección que altere la producción, el transporte o la liberación de AVP.
La AVP-R es el resultado de enfermedades que alteran la capacidad de los conductos colectores de los riñones para responder ante la AVP.
Tanto la AVP-D como la AVP-R se caracterizan por el deterioro de la reabsorción de agua renal, lo que da lugar a la producción de orina (poliuria) hipotónica (diluida) excesiva, en volúmenes que oscilan entre los 3 y los >20 litros por día. Esto va acompañado de una sed importante y un aumento de la ingesta de bebida (polidipsia), ya que la osmosensibilidad central y la barosensibilidad periférica hacen que la sed central y los comportamientos dependientes de la sed mantengan el volumen circulante y el estado osmolar.
Los pacientes con AVP-D o AVP-R por causa de una etiología no traumática generalmente presentan un inicio lento de los síntomas. Los pacientes con AVP-D después de una lesión cerebral traumática o una cirugía de la hipófisis suelen experimentar un inicio más rápido de los síntomas.
El embarazo se asocia a varios cambios en la regulación de la sal y el agua.[44] Se puede desarrollar una AVP-D transitoria como consecuencia de una disminución en el umbral osmótico para la sed y la liberación de AVP y una disminución en la osmolalidad plasmática. También aumenta 4 veces la depuración metabólica de AVP debido a la producción placentaria de la vasopresinasa/oxitocinasa. Además, el embarazo puede agravar la gravedad de cualquier AVP-D o AVP-R existente.[1][6][45]
Clasificación
Síndromes de poliuria-polidipsia
Polidipsia primaria:
Un síndrome clínico debido a la excesiva ingesta de líquidos con la subsiguiente poliuria. Por lo general, sugiere un trastorno de la sed sin alteración de la secreción de la acción de AVP.
Puede ser producida por fármacos que causan sequedad en la boca, estar asociada a síndromes psiquiátricos o ser habitual.[1]
AVP-D:
Se debe a la síntesis o liberación defectuosas de arginina vasopresina (AVP) desde el eje hipotalámico-hipofisario.[1]
Puede ser congénita (hereditaria) o adquirida (p. ej., procedimientos neuroquirúrgicos para tumores, traumatismos, enfermedades autoinmunitarias, infiltrantes, vasculares).[3][4]
AVP-R:
Se debe a una insensibilidad o resistencia renales a la AVP, que resulta en una falta de permeabilidad al agua de los conductos colectores.[1]
Puede ser congénita (hereditaria) o adquirida (p. ej., inducida por fármacos, en particular el litio; hipercalcemia, hipopotasemia; uropatía obstructiva).[5]
También se puede observar AVP-D o AVP-R transitoria en el embarazo (anteriormente conocida como diabetes insípida gestacional), debido al metabolismo acelerado de AVP por la producción placentaria de cisteína aminopeptidasas.[6]
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad