Epidemiología
El síndrome de Turner afecta de igual manera a todas las razas y regiones del mundo. La presentación es esporádica, excepto en casos raros en los que una deleción pequeña del cromosoma X puede pasar de madres a hijas.
El número de mujeres diagnosticadas prenatalmente puede aumentar con un mayor uso de pruebas prenatales no invasivas (NIPT).[8] De todos modos, es necesario un cariotipo posnatal para confirmar el diagnóstico.[5] Históricamente, menos del 10% de los casos se diagnostica antes del nacimiento; otro 20% se detecta en la lactancia debido a la presencia de linfedema, cuello alado o defectos cardíacos congénitos. Relativamente pocos pacientes son diagnosticados durante la primera infancia y suelen presentar una estatura baja. La mayor proporción de detección es entre los 10 y 16 años de edad, debido a una combinación de estatura baja marcada y retraso puberal. Otro 10% se diagnostica en la edad adulta, debido a la amenorrea secundaria. Europa y EE. UU. poseen perfiles de diagnóstico similares.[9][10]
[Figure caption and citation for the preceding image starts]: Datos de los perfiles de determinación del síndrome de Turner; 300 mujeres; edad diagnóstico, edad (en años) en el momento del diagnósticoDe la colección personal de la Dra. Carolyn Bondy, MS (National Institute of Child Health and Human Development natural history study 2001-2007; McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775) [Citation ends].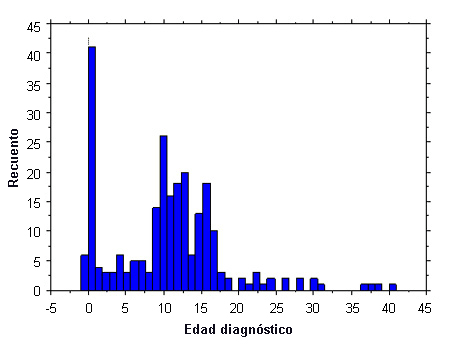
Mientras que el cribado citogenético a larga escala de recién nacidas llevado a cabo en las décadas de los 70 y los 80 indicaba una incidencia del síndrome de Turner de aproximadamente 1 caso por cada 2000-2500 nacimientos de niñas nacidas vivas,[11][12] los estudios más recientes sugieren una incidencia menor que es de 1 caso por cada 5000 niñas nacidas vivas debido a un diagnóstico prenatal asociado a la interrupción del embarazo.[13]
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad