Etiología
No se conocen factores causales o predisponentes para el síndrome de Turner. La presentación es esporádica, excepto en casos raros en los que una deleción pequeña del cromosoma X puede pasar de madres a hijas.
La pérdida de todo o una parte de un cromosoma sexual, X o Y, durante la embriogénesis temprana generalmente da lugar al desarrollo de un feto femenino con anomalías de desarrollo. La monosomía para el cromosoma Y es inviable. Los fetos 46,XY que pierden un cromosoma Y en una etapa del desarrollo relativamente tardía pueden contar con la cantidad suficiente de cromosomas Y como para conseguir un desarrollo masculino completo o parcial. Sin embargo, a estos pacientes se los identifica como masculinos y se consideran bajo el diagnóstico de trastornos de diferenciación sexual en vez del síndrome de Turner. Los hombres con estirpes celulares 45,X presentan grados variables de estatura baja, retraso puberal o infertilidad y riesgo de trastornos cardiovasculares.[14]
El fenotipo se determina según el momento de la pérdida cromosómica, la persistencia de estirpes celulares normales (abundancia relativa y distribución tisular) y los antecedentes genéticos.
Fisiopatología
Las regiones homólogas de los cromosomas sexuales (región seudoautosómica [PAR]) se comportan como autosomas en cuanto a que experimentan apareamiento y recombinación homólogos y contienen genes que escapan a la inactivación del cromosoma X. Se considera que estos genes son necesarios en dosificación doble tanto en hombres como en mujeres. Existen cerca de 20 genes anotados y varios genes más pronosticados en la PAR1, ubicada en el terminal Xp. En la PAR2, en el terminal Xq, se encuentra un grupo más pequeño de genes homólogos. En el síndrome de Turner está involucrada la haploinsuficiencia de los genes PAR1.
[Figure caption and citation for the preceding image starts]: Regiones pseudoautosómicas de los cromosomas X e YDe la colección personal de la Dra. Carolyn Bondy, MS [Citation ends].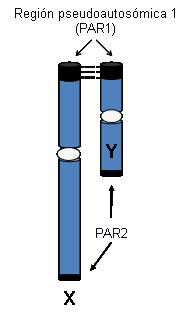
No se conoce bien la fisiopatología específica detrás de las anomalías de desarrollo observadas en el síndrome de Turner.
El crecimiento longitudinal de los huesos y el desarrollo esquelético alterados que dan lugar a la estatura baja se han relacionado con la haploinsuficiencia de un gen cromosómico sexual específico, el gen SHOX, ubicado en la PAR, el cual codifica un factor de transcripción que se expresa en el desarrollo de huesos y cartílagos.[15][16]
La haploinsuficiencia de otros genes pseudoautosómicos causa los defectos cardiovasculares congénitos que producen mortalidad elevada en el síndrome de Turner.[17][18] Dos genes, TIMP1 y TIMP3, que están ubicados en el brazo corto del cromosoma X cuando son hemicigotos, aumentan el riesgo de aortopatía con un odds ratio de 12.86.[19]
La causa de insuficiencia ovárica prematura sigue siendo dudosa, pero se ha implicado a los genes ubicados tanto en los brazos cortos como en los largos del cromosoma X.[20] El centro de inactivación del cromosoma X (CIX) está ubicado en el brazo largo, Xq. La deleción del Xq distal al centro de inactivación del cromosoma X se asocia con una insuficiencia ovárica prematura más que otras características del síndrome de Turner. Los ovarios se forman de manera normal en los fetos femeninos 45,X, aunque la mayoría presenta muerte acelerada del ovocito y degeneración ovárica en forma de estrías fibrosas.[21]
La haploinsuficiencia de los genes ubicados en Xp da lugar a rasgos neurocognitivos distintivos, como un rendimiento verbal mayor que el visual-espacial y dificultades para reconocer expresiones faciales.[22]
Clasificación
Clasificación citogenética[6]
45,X sin mosaicismo
Cromosoma X único en todas las células somáticas (monosomía X); afecta aproximadamente al 60% de las pacientes con síndrome de Turner.
X o Y fragmentado
Deleciones Xp (46,X,delXp), cromosomas isoXq (46,X,iXq), deleciones Xq y cromosomas X o Y en anillo con deleciones intersticiales sustanciales. El cromosoma isoXq, que proviene de la deleción de Xp y la fusión de 2 brazos largos, es la anomalía estructural más común asociada al síndrome de Turner. La deleción de una gran parte de Xp también se asocia con el síndrome. A menudo, el cromosoma sexual anómalo se pierde en algunas células durante el desarrollo embrionario, lo que da como resultado el mosaicismo de una estirpe celular 45, X, además de la línea X frágil, 46, X (ver en la figura A a continuación).
Mosaicismo 45,X
Existen dos tipos de mosaicismos.
La pérdida de un cromosoma sexual durante las divisiones celulares embrionarias tempranas puede provocar una combinación de células 46,XX normales y células 45,X en proporciones variables en los tejidos corporales e involucra a alrededor del 15% de las pacientes con síndrome de Turner: por ejemplo, 45,X (50%)/46,XX (50%) (ver en la figura B i. a continuación). La proporción relativa de células normales en diferentes tejidos influye en el fenotipo.
Por el contrario, la formación de un embrión 46,XabnX a menudo viene acompañada de la pérdida del cromosoma X fragmentado durante algunas divisiones celulares embrionarias, lo que da lugar al mosaicismo de una estirpe celular 45,X monosómica junto con una estirpe celular que contiene un cromosoma X fragmentado: por ejemplo, iXq (ver B ii. en la figura a continuación). Este individuo no presenta células normales; se espera el fenotipo completo.[Figure caption and citation for the preceding image starts]: Alteraciones del cromosoma X en el síndrome de Turner; ver explicación en el textoDe la colección personal de la Dra. Carolyn Bondy, MS [Citation ends].
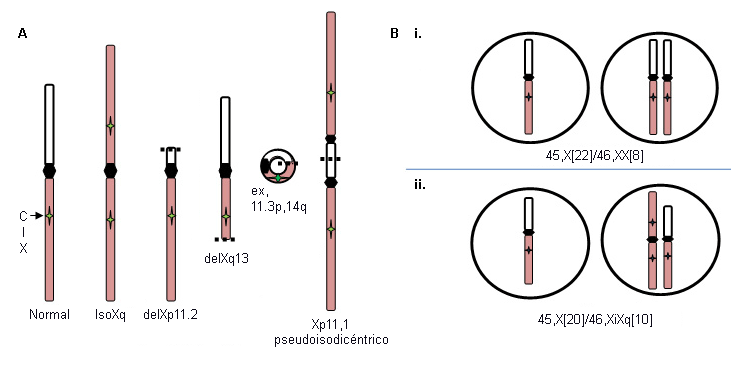
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad