Etiologia
A doença de Hirschsprung é uma doença genética com um padrão de herança complexo.[19][24] Estudos genéticos identificaram variantes em mais de 24 genes associados à doença.[1][26]
O gene rearranjado durante a transfecção (RET) é o gene mais comumente implicado na doença de Hirschsprung, com mutações encontradas em 50% dos casos familiares e 15% a 35% dos casos esporádicos.[19][27][28] As mutações em RET também estão implicadas na neoplasia endócrina múltipla do tipo 2 (MEN2), que também está associada à doença de Hirschsprung.[29] Consulte Síndromes de neoplasia endócrina múltipla.
O receptor de endotelina do tipo B (EDNRB) é o segundo gene mais comumente mutado na doença de Hirschsprung esporádica, com uma frequência de aproximadamente 5%.[24] Mutações no EDNRB levam à síndrome de Shah-Waardenburg, caracterizada por surdez congênita, anormalidades de pigmentação e doença de Hirschsprung.[30]
Outros genes implicados na doença de Hirschsprung incluem SOX10 e PHOX2B, que está associado à síndrome de Haddad; uma combinação da doença de Hirschsprung e da síndrome de hipoventilação central congênita.[31]
Fisiopatologia
A ausência de células ganglionares e a presença de nervos hipertróficos, bem como um aumento da enzima acetilcolinesterase, são as chaves para o diagnóstico patológico do segmento intestinal disfuncional.[32][33][34][35][Figure caption and citation for the preceding image starts]: Seção histológica, incluindo mucosa com submucosa do reto, mostrando agrupamentos de células ganglionares no plexo submucoso. Isto descarta a doença de Hirschsprung neste nívelCorman ML. Colon and rectal surgery. 5ª ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2005:555; usado com permissão [Citation ends].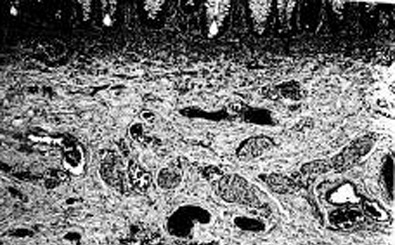 [Figure caption and citation for the preceding image starts]: Seção histológica, incluindo mucosa e a submucosa do reto, mostrando troncos nervosos tortuosos e hipertróficos do plexo submucoso. Não há evidências da presença de célula ganglionar. Isto estabelece o diagnóstico da doença de HirschsprungCorman ML. Colon and rectal surgery. 5ª ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2005:555; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Seção histológica, incluindo mucosa e a submucosa do reto, mostrando troncos nervosos tortuosos e hipertróficos do plexo submucoso. Não há evidências da presença de célula ganglionar. Isto estabelece o diagnóstico da doença de HirschsprungCorman ML. Colon and rectal surgery. 5ª ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2005:555; usado com permissão [Citation ends].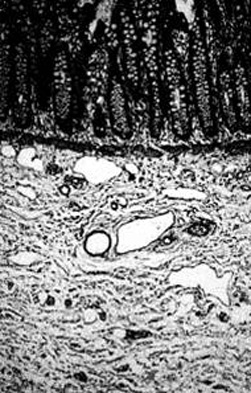
A coloração de patologia demonstra um aumento significativo do número de fibras nervosas hipertróficas localizadas na muscular da mucosa, na lâmina própria e na submucosa, e um aumento da atividade da acetilcolinesterase. Os nervos hipertróficos são aqueles que têm diâmetro maior que 40 mícrons.[35]
Acredita-se que a doença de Hirschsprung seja causada pelo desenvolvimento anormal do sistema nervoso entérico. Durante a embriogênese, parece haver uma parada na migração craniocaudal das células ganglionares neuroentéricas da crista neural para o trato gastrointestinal superior, descendo pelas fibras vagais e ao longo do intestino distal.[36] Acredita-se que isso seja resultado de mutações em genes como RET, GDNF, EDNRB, SOX10 e PHOX2B, que são essenciais para as vias de sinalização que regulam a migração dessas células durante o desenvolvimento embrionário.[1][37] Como consequência, faltam células ganglionares no plexo mioentérico, no plexo de Henle e no plexo de Meissner.[18] Acredita-se que o número e a gravidade das mutações, além de fatores epigenéticos e ambientais, influenciam o comprimento do segmento aganglionar.[1]
Sob circunstâncias normais, os gânglios parecem agir como um caminho final comum tanto para influências simpáticas como parassimpáticas. Sua ausência pode supostamente produzir contrações descoordenadas no intestino afetado. Espasmo, ausência de peristaltismo propulsor e contração maciça do segmento aganglionar foram bem documentados, em associação com a falta de relaxamento do intestino e espasmo do esfíncter interno.[38][39] Os resultados clínicos desses eventos fisiopatológicos são a obstrução colônica funcional parcial ou total.
O papel do óxido nítrico como um neurotransmissor responsável pela ação inibidora dos nervos entéricos intrínsecos está sendo elucidado .[40][41]
Classificação
Comprimento do segmento aganglionar
A doença de Hirschsprung pode ser categorizada pelo comprimento do segmento aganglionar. Isso determina a gravidade da doença e o tratamento subsequente.[4]
Segmento curto (retossigmoide)
O segmento aganglionar inclui o reto e grande parte do cólon sigmoide. Isso compreende 80% a 85% dos casos.[5]
Segmento longo
O segmento aganglionar se estende além do cólon descendente sigmoide (embora essa definição varie entre os estudos).[4][6] Pessoas com doença de segmento longo geralmente apresentam uma evolução mais complicada e necessitam de consideração particularmente cuidadosa em relação ao tratamento.[4][7] Muitas crianças com doença de segmento longo precisarão de desvio intestinal como parte do manejo inicial.[1] Isso compreende até 20% dos casos.[5]
Aganglionose colônica total (ACT)
Uma doença extremamente grave na qual o cólon inteiro é aganglionar, frequentemente incluindo um comprimento variável do íleo terminal. Isso compreende cerca de 8% dos casos e geralmente exigirá desvio intestinal seguido de proctocolectomia e reconstrução posteriormente na vida.[6][8]
Intestinal total
A aganglionose intestinal total é rara (<1% dos casos) e ocorre quando a quantidade total de intestino delgado ganglionar é menor que 40 cm.[9] O tratamento inclui reabilitação intestinal, nutrição parental e, às vezes, transplante intestinal.[10]
Segmento ultracurto
Há algum debate sobre a existência desse subtipo e uma revisão sistemática de 2021 do Comitê de Desfechos e Práticas Baseadas em Evidências da American Pediatric Surgical Association recomendou que esse termo não deveria ser usado.[4]
O uso deste conteúdo está sujeito ao nosso aviso legal