Etiologia
A exposição a corticosteroide exógeno é a causa mais comum de síndrome de Cushing.
A maior parte (70% a 80%) dos pacientes com síndrome de Cushing endógena apresenta adenomas hipofisários secretores de hormônio adrenocorticotrófico (ACTH); no entanto, apenas 10% de todos os adenomas hipofisários secretam uma quantidade excessiva de ACTH .[8][16] Adenomas corticotróficos silenciosos são imunopositivos para ACTH, mas aparecem clinicamente como adenomas não funcionais. No entanto, grandes adenomas raramente se tornam secretores de ACTH e causam doença de Cushing.[17] Descobriu-se um novo gene, protease 8 específica para ubiquitina, que é mutado na doença de Cushing.[18]
Cerca de 10% dos pacientes com síndrome de Cushing endógena apresentam adenomas adrenais com secreção não regulada de cortisol, mas apenas de 5% de todos os adenomas adrenais desenvolvem secreção autônoma de cortisol.[8][14] Mutações na via cAMP/PKA são a causa da maioria dos casos de hipercortisolismo adrenal primário.[19] Não foram identificados fatores de risco específicos ou modificáveis que causam hipercortisolismo endógeno. A etiologia de superprodução de ACTH do adenoma hipofisário e de superprodução de cortisol do adenoma adrenal é pouco compreendida.
Apenas cerca de 1% dos pacientes com síndrome de Cushing têm carcinoma adrenal. Por outro lado, a superprodução adrenal de cortisol é vista em 50% a 60% dos carcinomas adrenais, resultando em síndrome de Cushing independente de ACTH.[10]
Fisiopatologia
As manifestações clínicas resultam da exposição excessiva do tecido ao cortisol. O grau de manifestação dos sintomas é amplamente, se não inteiramente, baseado no nível de excesso de cortisol. Pacientes com hipercortisolismo leve a moderado geralmente possuem um fenótipo menos proeminente com intolerância à glicose, dislipidemia, doença osteometabólica e ganho de peso anormal, mas são difíceis de serem diferenciados de outros pacientes com a síndrome metabólica. À medida que o hipercortisolismo aumenta, as características físicas pioram, com o desenvolvimento de estrias, adiposidades supraclaviculares e fraqueza muscular proximal. A secreção do hormônio adrenocorticotrófico (ACTH) ectópico de tumores neuroendócrinos pode se manifestar como casos mais graves, com a apresentação abrupta dos sintomas e cortisol e ACTH muito elevados. Esses pacientes também podem apresentar fraqueza muscular grave e perda de peso.
Classificação
Etiologias de hipercortisolismo
Dependente de hormônio adrenocorticotrófico (ACTH)
Causado por doenças que apresentam níveis elevados ou anormais de ACTH estimulando a superprodução adrenal de cortisol.
Os adenomas hipofisários secretores de ACTH (doença de Cushing) e os tumores secretores de ACTH ectópico são duas formas da doença dependente do ACTH. Os tumores secretores de ACTH ectópico geralmente têm origem broncogênica ou neuroendócrina.
O hormônio liberador de corticotropina (CRH) ectópica pode ser considerado nesta categoria mas, por ser extremamente raro, não é especificamente discutido aqui.
Independente do ACTH
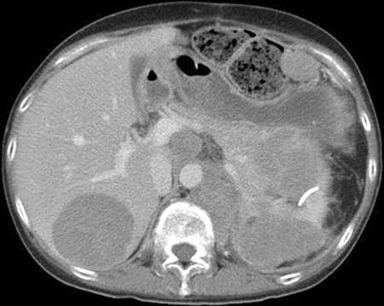
A síndrome de Cushing independente de ACTH é causada pela secreção excessiva de cortisol pelas glândulas adrenais, apesar de um nível de ACTH suprimido. Estão incluídos nesta categoria os adenomas adrenais, a hiperplasia adrenal bilateral e, raramente, o carcinoma adrenal. Carcinoma de adrenal é extremamente raro e normalmente se apresenta como uma massa adrenal grande (> 5 cm) e de crescimento rápido.[Figure caption and citation for the preceding image starts]: Exame abdominal computadorizado exibindo um tumor adrenocortical infiltrando o pâncreas e rim esquerdo e com metástase no fígado, baço e nódulos centraisFrom BMJ Case Reports 2010; doi:10.1136/bcr.07.2009.2100 [Citation ends].
A hiperplasia adrenal macronodular independente de ACTH (AIMAH) é uma forma rara da doença adrenal nodular bilateral. A fisiopatologia da hiperplasia adrenal macronodular independente de ACTH é a estimulação do receptor aberrante. Foram identificados pelo menos 10 diferentes receptores aberrantes que causam a hiperplasia adrenal macronodular independente de ACTH. Entre eles, o receptor do polipeptídeo inibitório gástrico, receptores beta-adrenérgicos, receptor de vasopressina, receptor de serotonina, receptor de angiotensina II, receptor de gonadotropina coriônica humana/hormônio luteinizante e receptor de leptina.[2] A estimulação desses receptores causa o crescimento inadequado de nódulos monoclonais e policlonais bilaterais grandes.[3][4] Clinicamente, a hiperplasia adrenal macronodular independente de ACTH se apresenta com mais frequência na quinta e sexta décadas de vida, com excesso de produção de glicocorticoide e mineralocorticoide.[5] Nas imagens, são observadas glândulas adrenais bastante aumentadas bilateralmente.
A doença adrenocortical nodular pigmentada primária (PPNAD), que frequentemente faz parte do complexo de Carney (uma síndrome de neoplasia endócrina múltipla caracterizada por lentigos, mixomas cardíacos e cutâneos e tumores endócrinos; pode ser herdada de forma autossômica dominante), é uma doença rara causada por vários nódulos adrenais pequenos, funcionando de forma autônoma, com tamanhos que variam de microscópico a 1 cm. No exame patológico, os nódulos têm pigmento escuro, e o córtex adrenal interveniente é atrófico. Acredita-se que as mutações da subunidade reguladora tipo 1 alfa da proteína quinase A (PRKAR1A) podem ser as mutações causadoras. A doença adrenocortical nodular pigmentada primária tem idade de distribuição bimodal, com a maioria dos casos diagnosticada na segunda e terceira décadas de vida.[6] Pacientes com PPNAD tendem a ser bastante magros e não apresentam a típica obesidade central vista em outras causas de síndrome de Cushing. Osteoporose grave, baixa estatura e grave atrofia muscular são características comumente presentes nos pacientes com doença adrenocortical nodular pigmentada primária.[7]
Exógena
Os pacientes que tomam corticosteroides exógenos, por qualquer motivo, podem desenvolver características da síndrome de Cushing. Quando doses elevadas de glicocorticoides são interrompidas, os pacientes podem desenvolver insuficiência adrenal, apesar de um fenótipo clínico da síndrome de Cushing.
O uso deste conteúdo está sujeito ao nosso aviso legal