Etiologia
Embora a EM tenha sido classicamente considerada uma doença da substância branca do sistema nervoso central, atualmente, há evidências substanciais que respaldam o envolvimento tanto da substância branca quanto da cinzenta.[16] Existem aparentemente componentes inflamatórios e degenerativos que podem ser desencadeados por um fator ou fatores ambientais em pessoas geneticamente suscetíveis.
Parentes em primeiro grau de pacientes com EM têm probabilidade 20-40 vezes maior de desenvolver a doença do que a população geral.[17][18] O risco ao longo da vida de acordo com a idade para crianças que têm um genitor com EM é de aproximadamente 2% a 3%.[19] A prevalência mundial de EM familiar foi estimada em 12.6%.[20] Embora a genética da EM seja multifatorial, é provável que os genes na região do antígeno leucocitário humano (HLA) e da região de interleucina estejam envolvidos.[21][22] Testes genéticos para prever riscos de EM não são recomendados atualmente.
Fatores ambientais que se tem afirmado estarem envolvidos na EM incluem toxinas, exposições virais e exposição à luz do sol (e seu efeito no metabolismo da vitamina D).[23][24] Embora alguns pesquisadores sugiram que a EM é causada por um vírus, nenhum dos quase 20 vírus considerados nos últimos 20 anos foi comprovado como causador. O vírus Epstein-Barr (EBV) é o vírus que demonstra maior relação com o aumento do risco de EM.[25][26] Há também sugestões de que o herpes-vírus humano tipo 6 esteja ligado à EM, mas isso não foi demonstrado de forma conclusiva.[27]
Às vezes, as recidivas são desencadeadas por infecções ou por alterações hormonais pós-parto. Algumas literaturas sugerem que traumas agudos ou eventos estressantes podem ser desencadeadores, embora isso seja controverso.[28][29]
Fisiopatologia
A patogênese exata da esclerose múltipla (EM) é desconhecida. Não há antígenos ou anticorpos específicos ou sensíveis, e há debates sobre se a EM representa uma doença única ou uma síndrome de subgrupos de pacientes patogenicamente heterogêneos. As conceitualizações da imunopatologia da EM envolvem 2 fases distintas, mas sobrepostas e relacionadas: a inflamatória e a degenerativa.
Durante o estágio inicial da fase inflamatória, os linfócitos com potencial encefalitogênico são ativados na periferia por fatores como infecção ou outro estresse metabólico. Essas células T ativadas procuram se infiltrar no sistema nervoso central (SNC) pela ligação a um receptor em células endoteliais. Essa interação, mediada pela produção de metaloproteinases da matriz, permite uma quebra na barreira hematoencefálica, ocasionando suprarregulação das moléculas de adesão endotelial e influxo adicional de células inflamatórias. As células T produzem citocinas inflamatórias que causam toxicidade direta e também atraem macrófagos que contribuem para a desmielinização. A disseminação de epítopos ocorre precocemente e contribui para a complexidade da imunopatologia.
Acredita-se que o componente degenerativo da EM reflita degeneração e perda axonais. A desmielinização prejudica o suporte axonal e provoca uma desestabilização dos potenciais da membrana axonal, que causa degeneração distal e retrógrada com o tempo. Há também uma sugestão de que células inflamatórias, anticorpos e complementos contribuam para a lesão axonal. Lesão axonal foi identificada em regiões de inflamação ativa, indicando que ela começa no início do processo da doença.[30]
Patologicamente, a EM caracteriza-se por áreas multifocais de desmielinização, perda de oligodendrócitos e astrogliose com perda de axônios principalmente na substância branca do SNC, embora as lesões corticais também possam ter um papel importante. A heterogeneidade clínica e os estudos de espécimes de biópsia e autópsia sugerem que os mecanismos que provocam danos ao tecido diferem de um paciente para outro.
A EM remitente recidivante mostra maior atividade inflamatória, seguida pela EM progressiva secundária. Acredita-se que a EM progressiva primária seja, sobretudo, um processo degenerativo, embora alguns pacientes tenham recidivas e/ou lesões realçadas. Todas as terapias modificadoras de doença atualmente aprovadas para a EM são mais ativas contra inflamações.
Acredita-se que recidivas agudas da EM com distúrbio da função do SNC, como visão ou mobilidade, sejam períodos de atividade inflamatória elevada do sistema imunológico e devam ser tratadas como tais.
A progressão clínica, como a perda gradual da capacidade de movimentar-se com o passar dos anos e/ou pior recuperação das recidivas, é considerada uma manifestação da inflamação de baixa potência crônica e contínua combinada com o processo degenerativo.
Manifestações cerebrais e medulares da inflamação na ressonância nuclear magnética (RNM) mostram lesões por contraste com edema limitado, ao passo que as manifestações do processo progressivo na RNM mostram atrofia e hipointensidade em T1 (ou buracos negros).
O tratamento da EM tenta reduzir o potencial de desencadear surtos de atividade inflamatória conhecidos como recorrências, bem como limitar a extensão delas. A prevenção ou redução da inflamação supostamente reduz a perda axonal cumulativa e a incapacidade no longo prazo.
Classificação
Classificação fenotípica[2][3]
Os fenótipos da EM incluem uma consideração da atividade (com base em eventos clínicos e nos achados de imagem) e da progressão da doença.
1. Doença recidivante
A síndrome clinicamente isolada (SCI) é uma síndrome bem definida, como neurite óptica, disfunção do tronco cerebral/cerebelo ou mielite parcial, que não atende aos critérios de disseminação no espaço e no tempo. É considerada parte do espectro da EM recidivante. A SCI pode ser ativa ou não ativa. Caso haja um evento clínico ou atividade radiológica (realce por gadolínio ou lesões novas/aumentadas em T2) subsequente após o evento inicial na SCI, ela se torna EM remitente recidivante.
A EM remitente recidivante (EMRR) requer evidências clínicas e/ou de ressonância nuclear magnética (RNM) da disseminação no espaço e no tempo. A EMRR é também caracterizada como ativa ou não ativa dentro de um período especificado (por exemplo, 6 meses, 1 ano). Como base, as avaliações da atividade da doença devem ser conduzidas pelo menos uma vez ao ano.
2. Doença progressiva
A doença progressiva, seja progressiva primária (acumulação progressiva de incapacidade desde o início) ou progressiva secundária (acumulação progressiva de incapacidade após um ciclo recidivante inicial), tem quatro possíveis subclassificações que levam em conta o nível de incapacidade:
Ativa e com progressão (o indivíduo teve um ataque e também está piorando gradualmente)
Ativa, mas sem progressão (por exemplo, o indivíduo teve um ataque dentro de um período especificado anteriormente [isto é, 1 ano, 2 anos])
Não ativa, mas com progressão (por exemplo, a velocidade de caminhada diminuiu)
Não ativa e sem progressão (doença estável).
Variantes e doenças relacionadas à EM
Síndrome clinicamente isolada (SCI) e/ou evento desmielinizante monossintomático:
A síndrome desmielinizante monofásica, que pode ou não evoluir para EM, tem a mesma demografia que a EMRR.
Os critérios de McDonald de 2017 estabelecem que, em pacientes com uma SCI típica e demonstração de disseminação no espaço por exame clínico ou RNM, a presença de bandas oligoclonais específicas do líquido cefalorraquidiano permite o diagnóstico de EM; lesões sintomáticas podem ser usadas para demonstrar a disseminação no espaço ou no tempo em pacientes com síndrome da medula espinhal, supratentorial ou infratentorial; e lesões corticais podem ser usadas para demonstrar a disseminação no espaço.[4]
Bandas oligoclonais específicas no líquido cefalorraquidiano (LCR) e a presença de lesões na RNM ponderada em T2 no momento do primeiro evento clínico foram identificadas como fatores de risco independentes para conversão para EMRR.
Vários ensaios terapêuticos indicaram retardo até a ocorrência de um segundo evento clínico quando os casos de SCI são tratados com terapias modificadoras da doença na EMRR. [
 ]
[ ]
]
[ ]
Síndrome radiologicamente isolada (SRI)
O termo SRI é usado para pacientes que apresentam anormalidades cerebrais na RNM sugestivas de EM, mas que não tiveram sintomas que sugiram um evento clínico.
Distúrbios do espectro da neuromielite óptica:
Os distúrbios do espectro da neuromielite óptica não são mais considerados uma variante da EM por causa de sua imunopatologia distinta, das características à RNM e das opções de tratamento.
Recidivas graves que podem ser devastadoras, envolvendo apenas os nervos ópticos e a medula espinhal.
Causa perda de visão em um ou ambos os olhos e/ou lesões necróticas longas na medula cervical e torácica ao longo de diversos segmentos, resultando em paraparesia grave ou quadriparesia que pode ocorrer ao longo de dias ou semanas.[5][Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética (RNM) da coluna cervical ilustrando distúrbio do espectro da neuromielite óptica. Níveis múltiplos extensos de envolvimento da medula espinhal cervical com edema e desmembramento da barreira hematoencefálica, conforme ilustrado pela imagem ponderada com contraste em T1 (à esquerda). A imagem ponderada em T2 (à direita) indica a extensão da anormalidade do sinal que pode se manifestar clinicamente como quadriparesia com espasticidade grave e dorDo acervo do Dr. Lael A. Stone [Citation ends].
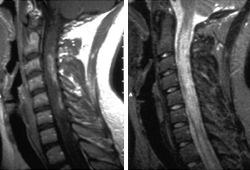
A demografia dos distúrbios do espectro da neuromielite óptica é diferente da EM típica, já que há uma predominância não branca.
O tratamento também difere, pois se trata de uma doença mediada por anticorpos que responde à imunossupressão e, em casos graves, à plasmaférese.[6]
É necessário testar a presença de autoanticorpos associados a distúrbios do espectro da neuromielite óptica (anticorpos antiaquaporina 4 ([AQP4]) e antiglicoproteína mielina-oligodendrócito [MOG]). Ensaios baseados em células são mais específicos que o ensaio de imunoadsorção enzimática (ELISA) e devem ser usados, se disponíveis.[7][8]
Encefalomielite disseminada aguda (EMDA):[9]
Doença monofásica que é uma entidade relacionada, porém distinta, da EM.
Apresenta-se com drástica disfunção do sistema nervoso central (SNC) pós-viral ou pós-vacinação, inclusive encefalopatia e múltiplas lesões cerebrais na RNM, que parecem ser simultâneas.
Mais comum na faixa etária pediátrica, mas pode ocorrer em adultos.
O episódio clínico pode durar de 3 a 6 meses.
Sem sinais, sintomas ou evidência radiográfica de dano prévio ou subsequente ao SNC.
O uso deste conteúdo está sujeito ao nosso aviso legal